Studying peripheral blood mononuclear cells (PBMCs) reveals cell-specific biomarkers—like target engagement and receptor occupancy—that improve decision-making in Phase I clinical trials. This makes these assays incredibly powerful tools in early-phase studies and has resulted in a growing trend of incorporating PBMC isolation and flow cytometry in Phase I clinical studies.
PBMCs are a group of white blood cells with a single nucleus, separated from whole blood through centrifugation. They include monocytes, lymphocytes, dendritic cells, T cells, B cells, and natural killer cells.
In this blog article, we’ll compare PBMCs and whole blood sample types and review their strategic applications in Phase I trials. We’ll also show how Celerion’s expertise in PBMC isolation and flow cytometry can accelerate your clinical program.
What Is Flow Cytometry?
Flow cytometry is a technique that measures cells in suspension. The cells are tagged with a fluorescent molecule that absorbs light at a specific wavelength and re-emits light at a longer wavelength. This can be leveraged to ‘gate’ or sort a specific cell population.
Why Choose PBMCs Over Whole Blood in Phase I Assays?
Whole blood contains red blood cells, PBMCs, platelets as well as hormones, lipids, proteins. While whole blood requires little initial sample handling compared to the PMBC isolation process. PBMC isolation can be advantageous, supporting several downstream analyses, such as ELISpot. In addition, the whole blood components may interfere with the assay and hinder stimulation or incubation of cells with targeted reagents, therefore PBMC isolation and appropriate cell culture environment are preferred.
Innovative Approaches to Clinical Pharmacology
Beyond immune characterization, PBMCs offer novel opportunities in clinical pharmacology. PBMC isolation and flow cytometry has traditionally been applied for immune cell characterization and biologics, however this procedure can also be leveraged for small molecules and for non-immunological purposes. In particular, PBMC isolation can support clinical pharmacology studies, for example:
Deeper Insights: PBMCs can provide deeper insights into cell-specific target engagement and intracellular drug activity, enhancing the understanding of drug mechanisms and effects.
Less Invasive Alternatives: PBMCs can potentially replace invasive procedures, such as skeletal muscle biopsies, by serving as surrogate biomarkers for tracking drug effects and activity.
Versatility: PBMCs can be leveraged for small molecules and non-immunological purposes in addition to biologic drug development, expanding their applicability in various clinical pharmacology studies.
Our PBMCs Experts are Ready to Support your Next Study
These approaches and their strategic applications were discussed early this year with industry peers at the ASCPT 2025 conference in Washington, DC, highlighting the growing role of PBMC-based assays in accelerating early-phase drug development.
Celerion has experience with incorporating PBMCs into a variety of clinical pharmacology studies including SAD/MAD, bioavailability, food effect, drug-drug interaction (DDI) and renal/hepatic impairment, as well as bioanalytical support for late phase studies. At Celerion, our expert clinical pharmacology & bioanalytical team can help unlock your next study by gaining deeper insights with PBMC & flow cytometry technology.
Weight reduction drugs are making a big splash! Since glucagon-like peptide 1 (GLP-1) receptor agonists were first approved for weight loss in 2021, there has been an estimated 700% increase in prescriptions in the US.1 This has led to a wave of new GLP-1 and incretin products entering into clinical research. Currently, there are nearly 150 novel GLP-1 receptor agonists in various stages of drug development globally.2 Beyond GLP-1, there are several other targets being explored for weight loss, also eager to swim in the same waters. In response, the Food and Drug Administration (FDA) recently updated guidance for industry to support drug development: Obesity and Overweight: Developing Drugs and Biological Products for Weight Reduction
The guidance addresses key aspects for drug developers, including recommendations for early and late phase trials, as well as input on sample size and primary endpoints. The following table highlights important study design elements.
As recently discussed in our blog article: Reframing the Definition of Obesity, the draft guidance also places emphasis on utilizing body mass index (BMI) for inclusion/exclusion criteria as well as the percent change in BMI for a primary endpoint. The FDA recognizes that while BMI is not a direct measure of adiposity (fat mass), it is a simple and effective assessment in which at least a 5% reduction is generally associated with improvement in metabolic and cardiovascular risk factors. In addition, to ensure the weight reduction effect is not due to loss of lean-body mass, body composition assessment by dual x-ray absorptiometry (DEXA) or another imaging modality is highly advised.
Safety Assessments for Weight Reduction Drugs
In terms of safety assessments, the FDA recommends monitoring changes in blood pressure and lipids. Early phase studies, such as SAD/MAD, also provide an opportunity to assess proarrhythmic risk (i.e., QTc prolongation), as well as immunogenicity potential for biologic therapies, including peptide drugs. The guidance also recommends including C-SSRS questionnaires for centrally acting drugs and echocardiographs for serotonin inhibitors.
Combination Products
Common adverse events associated with current GLP-1 therapies include nausea, vomiting, and muscle mass loss. To that end, an emerging trend is to optimize weight loss and minimize potential side effects with combination products. The guidance recommends assessing the safety and PK of each component in Phase I studies prior to initiating late-stage fixed-combination drug products trials.
Diving into Celerion’s Weight Reduction Experience
Celerion has extensive experience with a wide range of compounds for weight reduction, including GLP-1 receptor agonists, insulin sensitizers, and microbiota products. Our comprehensive experience with anti-obesity drugs covers all aspects of development; from first-in-human studies and proof-of-concept trials to clinical pharmacology studies to support labeling, such as drug-drug interaction – and bioavailability/bioequivalence studies.
Conclusion
As research in this indication continues, one can anticipate that the next generation of weight reduction therapies will render better safety profiles, more convenient drug administration (e.g., oral products or less frequent subcutaneous dosing), and improved patient adherence. Celerion is ready to help navigate the regulatory waters and support drug developers by leveraging our early phase clinical research experience, expertise, and efficiencies for smooth sailing ahead!
By Johannes Stanta, PhD, Global Scientific Director, Celerion Inc.
As drug development continues to evolve, few areas are advancing as disruptively as cell and gene therapies (CGTs). These highly complex and often personalized treatments are redefining what is possible in modern medicine. But while the science has leapt forward, many of the tools and assumptions behind therapeutic development have not. In particular, our continued reliance on animal models is becoming an increasingly obvious limitation.
That is where Model-Informed Drug Development (MIDD) comes in, and why now is the time to reexamine how we generate, interpret, and act on data throughout the development process.
Why Animal Models Fall Short for CGTs
CGTs operate through fundamentally different mechanisms than small molecules and biologics. Many of these therapies are intended for one-time administration, with long-lasting effects driven by genome editing, transgene expression, or cellular persistence. They do not follow classical dose-response relationships, do not exhibit linear clearance, and often behave in ways that are highly patient-specific and immune-mediated.
Despite this, drug developers and regulators continue to apply drug development frameworks designed for small molecules, relying on assumptions about repeated dosing, systemic pharmacokinetics, and linear modelling. This disconnect between therapeutic modality and development model often results in inefficiencies, suboptimal trial designs, and an overreliance on animal models that offer limited predictive value. Some regulatory agencies are beginning to acknowledge this gap. The FDA Modernization Act 2.0 and the agency’s 2024 roadmap to reduce animal testing both signal a shift toward more human-relevant, model-based approaches.
The Role of MIDD in CGT Development
MIDD integrates quantitative modelling, simulation, and data-driven decision-making across the product lifecycle. In the context of CGTs, this includes modelling the expansion, contraction, and persistence of engineered cells, predicting vector biodistribution across human tissues using physiologically based pharmacokinetic (PBPK) models, and understanding complex interactions between the therapy, disease pathways, and host immune responses through Quantitative Systems Pharmacology (QSP).
These tools make it possible to simulate hypothetical scenarios, define safe and effective first-in-human doses, and design more informative early-phase trials …well before administering a dose to a volunteer. As a result, MIDD reduces risk, improve administration of effective doses and decreases dependence on translation of animal-based safety and efficacy studies.
Bioanalysis: The Unsung Hero
Models are only as good as the data that inform them. Bioanalysis plays a crucial role in enabling model-informed development. Whether measuring vector DNA by PCR, quantifying cell expansion through flow cytometry, or assessing an expressed protein via ligand-binding assays, high-quality bioanalytical data are the foundation of any meaningful model.
Advanced bioanalytical platforms such as LC-MS/MS, immunoassays, digital droplet PCR, spectral flow cytometry and in vitrofunctional assays are not merely supportive tools, they are essential for the development and application of MIDD. Moreover, these human-relevant technologies are central to the FDA’s strategy for reducing animal testing. In the development of CGTs, mechanism-based bioanalysis is no longer optional. It is a scientific and regulatory necessity.
Looking Ahead: AI, NAMs, and a New Development Ecosystem
MIDD does not exist in isolation. It is now embedded in a broader framework that includes artificial intelligence (AI), New Approach Methodologies (NAMs), and growing regulatory support for model-based submissions. AI and machine learning are already being used to identify pharmacodynamic endpoints, biomarkers, stratify patients, generate virtual populations, and simulate clinical outcomes. At the same time, NAMs (including organ-on-chip systems and in vitro immune models) are producing more human-relevant preclinical data than traditional animal models.
When combined, these tools offer a smarter and more responsive way to develop CGTs. They support a more predictive understanding of efficacy and safety, reduce dependence on animal models that are often poorly translatable, and improve development timelines by avoiding lengthy and costly primate studies that yield limited actionable insights. In vitro methods can generate targeted, mechanistic data that feed directly into model frameworks, making them not only faster and more cost-effective but also better suited to the biology of CGTs.
Yet, development budgets are still heavily weighted toward animal testing, not because it delivers superior science, but because it remains deeply embedded in the regulatory process. As MIDD and NAMs continue to mature and regulatory standards emerge, this logic will be reversed. The focus will shift to the tools that provide the most decision-relevant, human-specific data.
The MIDD-driven ecosystem is also inherently more compatible with personalized medicine. CGTs are frequently developed for narrow patient populations or even individual patients. Traditional animal models are not equipped to handle this level of variability, whereas MIDD allows developers to model population-level and individual responses with far greater precision.
Final Thoughts
As someone working at the intersection of CGTs, bioanalysis, and model-based development, I have seen firsthand how these approaches are converging—and how rapidly expectations are changing across the industry. What was once aspirational is now an operational reality.
If we want to unlock the full potential of CGTs, we must move beyond legacy frameworks. MIDD, informed by robust analytics and bioanalytical data, offers a clearer, more efficient, and more ethical path forward. This is not only better science—it is better drug development.
If you are asking similar questions or actively working to reduce reliance on animal models through smarter, model-based development strategies, we would love to connect.
At Celerion, we believe that innovation in the lab shouldn’t come at the cost of simplicity or security. That’s why we’re excited to announce the latest evolution of LabNotes, our bioanalytical electronic laboratory notebook (ELN) software designed to make data management more intuitive and more powerful than ever before.
Whether you’re managing massive datasets or juggling multiple timelines, the newest version of LabNotes was built with one goal in mind: to help our clients move faster, think smarter, and protect their data with confidence.
What’s New in LabNotes?
The upgraded platform brings a host of improvements focused on speed, scalability, and security:
Streamlined Laboratory Documentation – Simplified workflows reduce documentation time and improve day-to-day efficiency.
Enhanced Dataset Management – Advanced tools make it easier to organize, analyze, and report on large data sets.
Improved Query Optimization – Faster, more reliable database access to keep projects moving forward without delays.
Advanced Security Features – With upgraded encryption and access controls, data protection is built into every layer.
Flexible Data Sharing – New archiving and extraction features make it easy to share information securely—without needing to install or run the core software.
Custom Report Integration – A newly added Report API allows teams to connect with third-party reporting tools for fully tailored data outputs.
Built for the Way You Work
“With this enhanced version, Celerion underscores its commitment to expanding the use of cutting-edge technology to support its clients in achieving business success.,” says Chad Briscoe, Executive Vice President of Global Bioanalytical Services at Celerion.“ These improvements demonstrate our continued commitment to innovation and excellence in all facets of our laboratory operations.”
“Our approach to software development is rooted in customer feedback and the latest industry trends. The updates in LabNotes empower clients with robust tools to manage and analyze their data effectively, enabling better decision-making and driving impactful business outcomes”, says Mark Williams, CEO of Terrington Data Management
by Johannes Stanta, PhD – Global Scientific Director, Sabina Paglialunga, PhD – Senior Scientific Director, and Aernout Van Haarst, PhD – Senior Scientific Director
The FDA’s recent decision to support non-animal methods for the safety evaluation of monoclonal antibodies (mAbs) is a significant—and long overdue—step forward in modernizing drug development. While the scientific community has long embraced the 3Rs framework (Refine, Reduce, Replace), regulatory acceptance has historically lagged behind. This has limited the practical application of advanced non-animal technologies beyond academic discovery and internal candidate selection.
Until now, animal testing has remained a regulatory default—often considered mandatory—with little room for alternative strategies. Good Laboratory Practice (GLP) enforcement has added further rigidity, making it challenging to integrate more flexible, human-relevant tools into regulated programs.
A Question of Translation, Not Tradition
The limitations of animal models are well established. Across therapeutic areas, particularly in immunology and oncology, the predictive value of animal studies for human outcomes is poor. In the context of mAbs, these limitations are especially pronounced. Safety signals often arise from excessive pharmacologic action or immune activation, rather than classical dose-dependent, off-target toxicity. These mechanisms are difficult—if not impossible—to model in animals, where interspecies differences in immune architecture obscure translatability.
This regulatory shift now enables the broader use of validated New Approach Methodologies (NAMs)—including organoid models, immune microphysiological systems, and in silico tools—as part of safety packages for investigational new drug (IND) applications. Notably, the FDA will also begin accepting real-world human safety data from other regulatory jurisdictions, providing an opportunity to reduce duplicative and ethically questionable animal studies.
Opportunities and Challenges for the Field
This shift raises important questions for the drug development ecosystem:
Can NAMs provide sufficient data to inform a safe starting dose for a first-in-human study? The current mAb paradigm relies on minimal anticipated biological effect level (MABEL) or physiologically active dose (PAD) to establish a first starting dose. While the MABEL approach can be fulfilled without animal testing as it often includes in vitro receptor occupancy assessments, the PAD approach tends to comprise of animal models, which may require rethinking or development of newer methodologies to substantiate a non-animal model.
How do we build confidence in the performance of NAMs? While early data on systems like liver chips and cytokine release assays are promising, widespread adoption will require validation, reproducibility, and clearly defined regulatory contexts of use.
Are current laboratory infrastructures ready to support the complexity of these models? Organoids and microphysiological systems demand expertise in cell biology, tissue engineering, and real-time functional readouts. Integrating such technologies into a GLP-aligned environment is a non-trivial task, especially as these concepts have often been deployed in non-GLP discovery or academic settings.
What frameworks are needed to standardize these methods across sponsors and regulators? Without harmonized protocols, the interpretation of NAM-based safety data risks inconsistency, delaying regulatory confidence.
How will bioanalytical and pharmacokinetic modeling capabilities evolve to complement these in vitro systems? Tools like physiology-based pharmacokinetic (PBPK) modeling, quantitative systems pharmacology (QSP), and immunogenicity prediction will need to be deeply integrated into the workflow and accelerated using machine learning approaches to continuously improve outcomes and applicability.
What type of research organizations will be best equipped to navigate this transition? It is reasonable to envision that GLP-accredited laboratories combining molecular and cellular assay expertise, regulatory bioanalysis and tight integration with clinical trial units may be best positioned to bridge nonclinical insights and clinical execution.
A Critical Inflection Point
The field has been scientifically prepared for this transition for years. Now, with regulatory momentum finally aligning, the challenge is operational. The focus must turn to building capacity, ensuring reproducibility, and developing harmonized protocols while educating regulators to work with these tools effectively.
This move by the FDA is not just a policy update—it is a call to action. If implemented thoughtfully, it will accelerate development timelines, reduce costs, uphold ethical standards, and, most importantly, improve the relevance of preclinical data to human biology.
The burden now shifts to the industry to answer: Are we ready to let go of legacy models and build a nonclinical paradigm that truly reflects human physiology?
by Chad Briscoe, Executive Vice President, Global Bioanalytical Services, Celerion
Someone whom I’ve worked with in the past and have had an opportunity to mentor in the bioanalytical field for some time recently asked me about the U.S. federal court ruling on LDTs. I realized I don’t really understand it as well as I would like to or as well as I should, and that I hadn’t been as “up-to-speed” on the current status of LDTs as used in the US or Europe. I decided to dig in and pull together a blog to help me learn and also help provide a guide for others that may want something that summarizes the current situation as I understand it.
In a landmark legal decision announced by the ACLA (American Clinical Laboratory Association) on March 31, a U.S. federal court vacated the FDA’s attempt to regulate Laboratory Developed Tests (LDTs) as medical devices. This preserved the long-standing CLIA oversight. Essentially preserving the status quo.
Meanwhile, across the Atlantic, the European Union’s In Vitro Diagnostic Regulation (IVDR) has been tightening control over diagnostic innovation. These two contrasting regulatory directions are reshaping the landscape for clinical and bioanalytical laboratories. Here’s my interpretation of what it means, why it matters, and how labs (in particular bioanalytical labs) can stay ahead.
Laboratory-Developed Tests (LDTs) have become foundational to the rapid evolution of diagnostic science. In recent years, due to the increasing complexity of LDTs and the focus on biomarkers in traditional bioanalytical labs, this has become important to laboratories beyond just the traditional Clinical Laboratories, central laboratories, and local hospital labs. These custom assays—designed, manufactured, and used within a single or small number of laboratories—often address unmet clinical needs, are used to support clinical trials and new treatment options. These have become key tools in affordable precision medicine. However, their regulation has been in flux for over a decade, driven largely by activities in the United States and European Union.
In the U.S., a federal court recently vacated the FDA’s 2024 final rule that sought to classify LDTs as medical devices. This decision, brought forward by the American Clinical Laboratory Association (ACLA), reasserts CLIA (Clinical Laboratory Improvement Amendments) as the primary regulatory authority over LDTs. In Europe, however, the new In Vitro Diagnostic Regulation (IVDR) has taken the opposite approach, intensifying regulatory oversight and requiring an onerous approval process with extensive documentation, validation, and risk classification for all diagnostic tests—including in-house laboratory tests.
Under the previous regulation, about 20% of laboratory tests were required to follow this path, but this has now increased to about 80% under IVDR.
Implications of FDA Regulating LDTs (If ACLA lawsuit had failed)
The FDA’s now-overturned rule would have subjected LDTs to the same premarket review, labeling, and post-market surveillance required for commercial in vitro diagnostic devices, developed for a mass market. Critics, including ACLA and the Association for Molecular Pathology (AMP), argued that this overstepped statutory bounds, stifled innovation, and jeopardized access to critical diagnostics.
The court’s ruling provides:
Regulatory Stability: Labs may continue operating under CLIA without additional FDA burdens.
Innovation Breathing Room: LDTs can still be developed and deployed quickly, often in response to urgent or rare clinical needs.
Continued Responsibility: While FDA oversight is paused, labs must continue to maintain rigorous internal validation practices and high-quality standards.
For bioanalytical labs, this ruling helps sustain agile assay development, enabling continued support for clinical trials, specialty testing, patient stratification, patient enrollment, and companion diagnostics.
The EU’s IVDR, which came fully into effect in May 2022, represents a complete overhaul of how diagnostics are regulated. Replacing the former IVDD, the IVDR imposes:
Risk-Based Classification (Class A to D) – though most LDTs are highly complex and therefore high risk
Performance Evaluation Requirements – requiring navigating a complex network of national Notified Bodies with varying interpretations of IVDR
Strict Use Conditions for In-House Tests – Typically, only hospital laboratories qualify when the test is used within the hospital.
Mandatory Quality Management Systems – ISO15189 is suggested, but accreditation bodies struggle to accredit non-routine clinical laboratories
Market Justification for Each In-House Test – requirement for regulatory review for each assay by a Notified Body
In stark contrast to the U.S. model, the IVDR now requires labs to prove that no CE-marked equivalent exists before using an LDT. Labs must also implement formal procedures for ongoing safety, performance monitoring, and clinical evidence gathering.
Key Differences at a Glance
For labs operating in clinical trials, translational science, or patient diagnostics, these changes affect everything from compliance planning to assay design and budgeting.
In the U.S.:
Labs retain operational flexibility.
No new device-level regulatory filings required.
Opportunity for rapid test iteration and clinical trial support.
In the EU:
Labs face new regulatory hurdles.
In-house test development is resource-intensive.
There may be delays or discontinuation of certain novel assays due to compliance costs.
Patient stratification for clinical trials will be reduced or stopped unless absolutely necessary.
Multinational labs performing LDTs must now manage dual compliance strategies:
In the U.S., keep internal systems CLIA-compliant while monitoring the policy landscape (e.g., VALID Act proposals).
In the EU, prepare for full IVDR implementation—especially if supporting EU-based clinical trials or patient testing.
Labs performing cross-border testing (e.g., EU patients using U.S.-based services) will need to harmonize quality, documentation, and reporting standards to remain compliant with IVDR, even if physically located outside the EU. Laboratories outside the EU struggle to find the correct national Notified Body that will review and approve their IVD assay.
The divergence between U.S. and EU regulation poses challenges—but also raises important questions:
Can future legislation, such as the VALID Act in the U.S., find a middle ground between oversight and innovation?
Will the EU refine or delay IVDR enforcement based on implementation feedback?
How can international standards be aligned to ease burdens for global labs?
Industry advocacy groups like ACLA and AMP are already working toward legislative solutions, while European regulators are issuing guidance to clarify IVDR expectations. Collaboration across borders will be key to future success.
Whether you’re a clinical lab director, regulatory affairs lead, or scientist in a bioanalytical CRO, the message is clear: the regulatory environment for diagnostics is evolving—rapidly and asymmetrically.
• In the U.S., the recent court decision protects flexibility and self-regulation—for now.
• In Europe, IVDR brings new rigor and central oversight—but also risks of reduced innovation.
• Globally, labs must adapt or risk falling behind.
As science continues to evolve and offers the opportunity for personalized medicine, regulatory oversight shouldn’t stand in the way of enabling it; instead, it should find a way to accelerate the safe implementation. In our industry, public health regulations that enable advanced clinical trial designs and early patient access to innovative new medicines are just as critical as scientific advancement.
Now is the time for bioanalytical labs to strengthen their quality systems, shake the fear of innovative solutions, and advocate for smart, science-driven oversight that puts patients first.
Two notable guidance documents addressing obesity and weight loss were issued in January 2025. Surprisingly, they have differing opinions on the role body mass index (BMI) plays in the definition of obesity and the clinical management of patients with obesity.
The recent FDA guidance defines obesity as a chronic disease characterized by excess adiposity and recommends using BMI, an anthropometric index, to classify weight groups. BMI is calculated as:
BMI = weight (kg) / [height (m)]2
The formula has been around since the 1800’s. It was developed by Adolphe Quetelet, a Belgian statistician, mathematician, and astronomer, in 1832. However, it wasn’t validated as a measure of obesity until the 1970s by physiologist Ancel Keys (reviewed in Pray & Riskin). Now, BMI is commonly applied to assess health and stratify disease risk. BMI cutoff values are applied to classify overweight (25-29.9 kg/m2) and obesity class 1 (30-34.9 kg/m2), obesity class 2 (35-39.9 kg/m2) and extreme obesity (≥40 kg/m2).
Weighing the Utility of BMI
While BMI is a convenient and long-standing measure to assess obesity, it does not differentiate between fat and lean muscle mass. It can either under- or over-estimate adiposity (fat mass). For example, older adults, conditions associated with bone or muscle mass loss, and certain ethnicities are prone to underestimation of obesity and fat mass by BMI, while conversely athletes could have overestimated obesity rates. Therefore, the Lancet Commission recommends that excess adiposity should be confirmed by either of the following as a second measure of fat mass, in addition to BMI:
Waist circumference
Waist-to-hip ratio
Waist-to-height ratio
Bioimpedance
Direct measures of body fat such as dual X-ray absorptiometry (DEXA) or magnetic resonance imaging (MRI)
One exception, however, is that clinicians may assume that a patient with a BMI ≥40 kg/m2 displays excess adiposity. The arguments to utilize or limit BMI in determining obesity are as follows.
Redefining Obesity
More poignant, the Lancet report also provides an updated and evidence-based definition of obesity, applying clinical and biological criteria for the diagnosis of this chronic illness.
Clinical obesity: a chronic, systemic illness characterized by alterations in the function of tissues, organs, the entire individual, or a combination thereof due to excess adiposity. Clinical obesity can lead to severe end-organ damage, causing life-altering and potentially life-threatening complications (e.g., heart attack, stroke, and renal failure).
Preclinical obesity: a state of excess adiposity with preserved function of other tissues and organs and a varying, but generally increased, risk of developing clinical obesity and several other non-communicable diseases (e.g., type 2 diabetes, cardiovascular disease, certain types of cancer, and mental disorders).
The aim of reframing the definition of obesity and its assessment is to encourage more accessibility and effective management for those with an unmet need. Until recently, obesity alone, without the presence of other diseases, was not considered a disease in itself (reviewed in Rubino et al.). This potentially led to negative implications for treatment options and insurance coverage for those with excess body fat without other comorbidities. However, relying on BMI alone with these new definitions could lead to overdiagnosis of obesity, therefore direct excess fat assessment in addition to physical work-up is recommended.
Conclusion
While these two reports may seem at odds with each other, the Lancet Commission does concede that BMI should be used only as a surrogate measure of health risk at a population level, for epidemiological studies, or screening purposes. The latter is most notable for drug developers. Therefore, in line with the FDA guidance, BMI will continue to be the main inclusion criteria for weight loss clinical trials. However, additional markers of excess fat could provide better insight into drug effects on adiposity. To that end, Celerion has extensive experience with weight reduction drugs, including GLP-1 receptor agonists, insulin-sensitizing drugs, and microbiota products. We also offer a full range of adiposity assessments, including BMI, body weight, waist circumference, and bioimpedance, as well as sophisticated imaging assessments such as DEXA, MRI, and FibroScan® (liver fat content).
Reference
FDA. Obesity and Overweight: Developing Drugs and Biological Products for Weight Reduction Guidance for Industry. 2025. https://www.fda.gov/media/71252/download
Pray R, Riskin S. The History and Faults of the Body Mass Index and Where to Look Next: A Literature Review. Cureus. 2023 Nov 3;15(11):e48230. DOI: 10.7759/cureus.48230
Rubino F et al. Redefining obesity: advancing care for better lives. The Lancet Diabetes & Endocrinology. 2025 Jan 14;13(2):75. DOI: 10.1016/S2213-8587(25)00004-X
By Sabina Paglialunga, PhD & Aernout van Haarst, PhD Senior Directors Scientific Affairs
Glucagon-Like Peptide-1 (GLP-1) receptor agonists first came to the market in 2005 as a type 2 diabetes mellitus treatment, and they have been making headlines again for their weight reduction effects. Specifically, semaglutide and tirzepatide, along with diet and exercise, are indicated for weight loss and have been shown to reduce body weight by up to 20%. GLP-1 receptor agonists reduce body weight by decreasing food intake, improving insulin sensitivity as well as delaying gastric emptying. The latter contributes to a longer feeling of fullness.
There are several techniques to assess gastric emptying in a clinical setting (Table 1). While scintigraphy is considered the gold standard for diagnostic purposes (e.g. for gastroparesis), an acetaminophen absorption assay applies classic pharmacology study designs to evaluate the role of gastric emptying in a drug-drug interaction study. In addition, the acetaminophen assay could be considered as a better predictor of drug absorption than the other approaches. In this assay, healthy volunteers consume acetaminophen dissolved in yogurt after administration of either a single dose or multiple doses (steady-state) of a drug that putatively alters gastric emptying. Then, changes in the acetaminophen PK profile are compared to a baseline or placebo condition.
When gauging acetaminophen absorption effects across various marketed GLP-1 receptor agonists, the overall acetaminophen maximum plasma concentration (Cmax) was reduced by 13-56%, a hallmark of delayed gastric emptying (Table 2). Interestingly, the GLP-1 receptor agonist, dulaglutide, and the dual GLP-1 and GIP receptor agonist, tirzepatide, are both characterized by a strong delayed gastric emptying upon a single dose administration, but the effect tends to subside as gastric emptying tachyphylaxis is observed after several weeks of treatment. Nonetheless, the tirzepatide effect of delayed gastric emptying was found to significantly reduce oral contraceptive PK by ~ 60% and, thus, the drug label advises patients using oral hormonal contraceptives to switch to or add a non-oral contraceptive method when initiating tirzepatide and after each dose escalation.
Biopharmaceutics Classification System (BCS) and Delayed Gastric Emptying
The rate of drug absorption depends on the solubility and permeability of an orally administered product. Solubility refers to the dissolution of the drug in an aqueous media, whereas permeability is the ability of a drug to cross membrane barriers and enter into the bloodstream. Oral small molecules, therefore, fall into one of four BCS categories depending on their solubility and permeability characteristics (Table 3). Delayed gastric emptying and reduced gastrointestinal (GI) motility can have different effects on drug PK dependent upon the drug’s BCS category. For instance, Class I drugs like acetaminophen display reduced Cmax in response to delayed gastric emptying. On the other hand, increased residence in the stomach leads to a higher degree of dissolved drug available for absorption immediately after entering the small intestine, resulting in increased PK exposure and delayed time to maximum concentration (Tmax) for Class II drugs. Reduced Cmax and delayed Tmax are anticipated for Class III drugs since intestinal permeability is rate-limiting. Finally, the anticipated PK changes due to delayed gastric emptying depend on whether solubility or permeability is the overall rate-limiting step in absorption.
A clinically relevant effect will depend on the extent of PK changes and the therapeutic index of the co-administered oral drug. For instance, while many of the GLP-1 drug interactions investigated to date may be ‘statistically significant,’ no dose adjustment is required because they were not found to be ‘clinically relevant.’ The one exception is tirzepatide, as described above. Nonetheless, for new GLP-1 drugs in development, it is imperative to evaluate potential drug interactions since this drug class can alter the efficacy of concomitant medications or increase the risk of adverse events as a result of higher exposure to a co-administrated drug.
Conclusion
GLP-1 receptor agonists have been known for their potential to delay gastric emptying. Consequent changes in AUC, Cmax, and Tmax values of orally administered concomitant medications may occur, which could impact drug efficacy or safety. Therefore, if a new oral drug in development is intended to be co-administered with a GLP-1 agonist, we highly recommend conducting a healthy volunteer drug-drug interaction prior to Phase 3 trials to characterize the impact on the PK of the candidate new drug. Similarly, novel GLP-1 receptor agonist-drug interaction studies evaluating the effect of delayed gastric emptying on the PK of oral medications from each BCS class should be considered.
By Sabina Paglialunga, PhD & Aernout van Haarst, PhD; Senior Directors Scientific Affairs, Celerion
Traditional drugs to treat cancer include chemotherapies, which are cytotoxic agents that non-specifically interrupt cell growth. While chemotherapy drugs effectively obstruct tumor development and growth, they also affect healthy cells, which can result in serious side effects as well as additional malignancies. Over the past two decades, alternative targeted therapies have emerged as less toxic options. Specifically, these drugs are designed to target and inhibit distinct molecular pathways involved in cancer cell growth and survival, such as tyrosine, serine, or threonine kinases. These enzymes and their receptors tend to be overexpressed in tumor cells and are involved in tumor cell proliferation, migration, and angiogenesis.
Compared to conventional chemotherapies, kinase inhibitors are generally considered non-cytotoxic compounds and, therefore, can be administered to healthy volunteers, especially for early-phase clinical pharmacology studies. This approach not only de-risks clinical investigations in patients but can also accelerate drug development. Robust safety and pharmacokinetic (PK) data can be acquired from healthy volunteers for first-in-human (FIH), food effect, and drug-drug interaction (DDI) studies with efficiency, quality, and swiftness.
Advantages of Healthy Volunteers over Patients:
More resilient human population if adverse events (AEs) occur:
AEs tend to be transient in nature, and healthy volunteers recover faster than patients do
Potential for less variable data
Healthy volunteers are not confounded by comorbidities that could result in data variability
Healthy volunteers are easier to recruit than most patient populations:
During early phase development, when efficacy has yet to be established, there is no potential benefit for patients, which may impact motivation to participate in a clinical trial
Patients are a more fragile population:
Due to polypharmacy, there may be potential drug interactions
Effect of disease on AE profiles:
In patient studies, care must be taken to distinguish drug-related AEs vs natural disease progression
Time and cost savings:
Development costs can be substantial as patient studies tend to require multiple sites, and slower recruitment can impact timelines
In general, unless the investigational compound causes direct DNA damage, regulatory agencies typically allow the administration of healthy volunteers. Overall, tyrosine kinase inhibitors tend to have an acceptable safety profile that justifies administration in healthy subjects. Nonetheless, they are known to elicit certain adverse effects, such as skin reactions, hepatotoxicity, and cardiovascular and gastrointestinal side effects, yet several mitigation steps can be taken to prevent or overcome these common class AEs.
Product Labeling Studies to Support Regulatory Submission
GI effects are quite common with kinase inhibitors; many cancer patients take acid-reducing agents (ARA) to help manage these side effects. The prevalence of ARA usage ranges between 20-70%, depending upon the cancer type, with GI and pancreatic cancers being the most widespread for ARA treatment. This is relevant for drug developers because of potential drug interactions between the kinase inhibitor and ARAs. In general, ARAs raise the stomach pH, making it a less acidic environment; this could reduce or increase drug bioavailability, which in turn could decrease efficacy or compromise safety, respectively.
Recent FDA guidance provides a physiochemical framework when an ARA-drug interaction study is recommended. Proton-pump inhibitors (PPIs) are the class of ARAs typically evaluated in such DDI studies as they are considered a worst-case scenario due to their relatively strong and long-standing effects on stomach pH. Typically, these PPI studies enroll healthy volunteers and can be combined with a non-medicinal ARA (e.g. orange juice, coke) or food effect arm in a single study design to maximize data output and understand how ‘real word’ factors will impact drug PK.
In addition to ARA DDI trials, other “drug labeling studies” that can be conducted in healthy volunteers include ADME, bioavailability(BA) / bioequivalent (BE), food effect, DDIs, and cardiac safety (TQT) assessments. These studies can support drug label claims and regulatory submissions. Conducting these studies in healthy volunteers allows for robust, high-quality data to be captured quickly.
Conclusions
In general, healthy, normal subjects can be considered for non-cytotoxic, small-molecule clinical trials, such as FIH, ADME, food effect, DDI, and cardiac safety (TQT) studies. Overall, Celerion has conducted over 140 clinical trials with kinase inhibitors since 2010. Relying on our extensive experience, we know how specific risks can best be mitigated. By leveraging data quality and saving time and costs, healthy volunteer oncology studies play a crucial role in accelerating cancer drug development, ensuring that only the most promising candidates proceed to patient populations at the safest dosing regimen.
By Sabina Paglialunga, PhD & Aernout van Haarst, PhD; Senior Directors Scientific Affairs, Celerion
Biologic drugs are pharmaceutical products derived from living organisms or their components. They represent a wide range of therapeutic treatments that include monoclonal antibodies, proteins, peptides as well as cell and gene therapies. Oligonucleotide therapeutics, on the other hand, may in principle be synthetic drugs, but by targeting specific RNA sequences to alter RNA and/or protein expression, they share certain features of true biologics. Generally, biologics and oligonucleotide drugs offer several advantages over traditional small molecule products such as targeted therapy, longer half-life (which can be associated with less frequent dosing leading to greater patient adherence) and even higher potency resulting in greater effectiveness. However, unlike most small molecules, plasma pharmacokinetic (PK) profiles of biologics and oligonucleotides may not reflect the target tissue distribution therefore, in some cases appropriate biomarkers or measures of target engagement may need to be assessed as an equivocal dose-effect relationship.
With biologics and oligonucleotide drugs playing an important role in modern medicine, over the past few years the FDA has issued updated guidance for oligonucleotides (draft 2022), peptides (draft 2023) and antibody drug conjugates (ADC; final 2024) to further promote these areas of drug development. Notably, many of the clinical pharmacology studies recommended to support small molecule regulatory submission, as well as efficiencies in corresponding study designs, may also pertain to biologics and oligonucleotide drugs. For example, if safe to do so, enrollment of healthy volunteers (HV) can expedite drug development, as this group is faster to recruit, associated with less variability in PK data, and have no confounding co-morbidities or concomitant medications compared to patients. The below table highlights which key clinical pharmacology studies may be recommended for each drug type.
Table: Key Clinical Pharmacology Studies to Support Drug Regulatory Submission
Product Labeling Clinical Pharmacology Studies
While large biologic drugs such as monoclonal antibodies and proteins are exempt from cardiac proarrhythmia risk assessment, a dedicated thorough QT (TQT) study may be recommended for peptide and oligonucleotide drugs, especially if a TQT substitution request via IHC E14 Q&A 5.1 or 6.1 is not sought.
A mass balance study employing a radiolabel is typically recommended for small molecules to understand and track adsorption, distribution, metabolism and elimination (ADME) of the parent drug. While not necessary for biologic and oligonucleotide drugs, there may be cases where an ADME study could be beneficial for peptide products, especially if their distribution and elimination pathways are unknown. The ADME study could also help inform the necessity of organ insufficiency PK studies, such as renal or hepatic impairment studies. For instance, if a protein or peptide drug is < 69 kDa, meaning small enough to be filtered by the kidneys, a renal impairment study is recommended. Similarly, a renal impairment PK study is recommended for oligonucleotide drugs if the therapeutic product is substantially eliminated by the kidneys. In addition, if the oligonucleotide drug targets the liver as part of its mechanism of action, a hepatic impairment PK study should be conducted. Peptides tend to be rapidly degraded by proteases and peptidases, bypassing hepatic elimination, thus negating the need for a hepatic impairment study. However, the recent draft FDA guidance document on peptide drug development does recommend a hepatic impairment PK study under certain conditions, such as:
The peptide drug is anticipated to undergo substantial hepatic metabolism or biliary excretion
The (lipid-conjugated) peptide drug is highly bound to serum albumin
The peptide drug’s pharmacological activity affects normal liver function
Finally, a drug-drug interaction (DDI) study may be recommended if the biologic or oligonucleotide product is a CYP or transporter substrate or modulator, or if a PD interaction with a concomitant medication is anticipated. For example, glucagon like protein-1 (GLP-1) analogs may delay gastric emptying, thereby affecting the absorption of co-administered treatments. In addition, therapeutic proteins that are proinflammatory cytokines or up/down-regulate cytokines levels may also need to be evaluated for DDI potential.
Special Considerations for ADC Drug Development
ADCs combine both small molecule and biologic drug components. The small molecule, often referred to as a ‘payload’, is conjugated to an antibody or an antibody fragment via a chemical linker. The antibody portion of the drug directs the payload to a specific tissue or target cell. Due to the combination of small molecule and biologic drug aspects, clinical pharmacology studies may be recommended to assess the unconjugated payload as well as the ADC or the total antibody, as necessary.
Conclusion
Clinical pharmacology studies such as TQT/cardiac safety, ADME, renal & hepatic impairment or DDI trials may be recommended for biologic and oligonucleotide drug development, depending on the type, size, PK profile and/or PD effects of the product. Celerion’s experienced team of scientific and operational experts are ready to support your biologic drug development program with efficient protocol design, effective study conduct and reliable data management & analysis.
By Sabina Paglialunga, PhD Director Scientific Affairs, Celerion
Need to run a renal impairment pharmacokinetic (PK) study, but don’t know where to begin? We have you covered! Celerion has managed more than 30 renal impairment PK studies over the past decade. We have a network of clinical sites and access to patients. To begin, we’ll guide you through 12 key questions to optimize study design.
Safety First
The first set of questions (Q1&2) relates to the safety profile of the drug in development. Is the investigational product safe to administer to patients with kidney dysfunction? If the study drug exacerbates kidney dysfunction, then it may not be suitable to dose in renal impaired participants. Next, we should consider the therapeutic range, as an increase in adverse events (AEs) and safety concerns may arise in patients with altered kidney function if the range is narrow.
Design Foundations
Assuming we are in the clear on these two fronts, we can then address the type of study needed (Q3&4). The FDA guidance, refers to full and reduced study designs. A full study explores the spectrum of organ dysfunction with cohorts ranging from normal → mild → moderate → severe, and in some cases kidney failure. A reduced study examines both ends of the continuum to compare normal vs severe conditions. If the investigational drug is mainly eliminated via the kidneys (i.e. renal clearance is > 10-20%) and/or is intended for patients with chronic kidney disease (CKD), then a full study is typically recommended. A reduced design is often sought when the investigational drug is likely to be administered to CKD patients yet the study drug is predominantly eliminated via the hepatobiliary route.
A hemodialysis study (Q5) may be recommended if the study drug is likely to be used in patients undergoing dialysis; the drug is not highly bound to plasma protein, and is small enough to escape dialysis filtering. In this case, the PK of the drug and any active metabolites are evaluated in patients both on- and off-dialysis days.
The dose regimen will depend on the PK characteristics of the study drug (Q6&7). A single dose may be administered if the drug exhibits dose-proportionality and displays time-independent PK. Multiple dose administration is often recommended when the drug or active metabolites show dose- or time-dependent PK characteristics.
Sample Collection
The following series of questions (Q8-10) help define the study sample schedule, collection, and analysis. In general, for renal impairment PK studies, blood samples are collected out to at least 3x the drug half-life. Metabolites representing >10% of total drug concentration should be measured in addition to the parent drug. If there is significant plasma protein binding, the unbound drug concentration should also be analyzed. Plasma protein binding is often altered in patients with renal impairment. Per the FDA guidance, a limited number of unbound samples can be measured if binding is not concentration- or time-dependent. In this situation, we recommend to collect at least 2 time points per participant; one at baseline and one at Tmax.
Cohort Size
The next question to consider is the number of patients per cohort (Q11). The FDA guidance recommends a powered study and sample size justification based on PK variability. Depending on drug variability (interCV%), 9-14 patients per cohort may be recommended.
Normal control matching is another question that regularly comes up during renal impairment study design discussions (Q12). We typically recommend the healthy control group to match patients by age (± 10 years), BMI (± 20%) and gender. There are two strategies, individual and mean matching. Individual or 1-to-1 matching allows for parallel patient and control enrollment, but can be difficult if a patient has uncommon characteristic. In some cases, a healthy control can match to more than one patient from different disease stage cohorts. With mean matching, the average values for the patient group are matched to the control subjects. This approach requires fewer subjects, but must wait until patient enrollment is complete.
Ready to Start
With this information in hand, our team of operational and scientific experts can propose the ideal number of sites, recruitment timelines, and other study design considerations. Rely on Celerion for budget-friendly and streamlined processes that leverage our long-standing and established relationships with key renal impairment PK Principal Investigators.
Adaptive clinical trials are characterized by innovative and flexible designs that incorporate safety and other information acquired during a study to instruct the next steps in a trial. Typically, these pre-planned adjustments include sample size refinement, subsequent dose level selection, or allocation ratio. In the case of patient studies, adaptive elements may consist of inclusion/exclusion criteria adjustment or stopping rules based on success or lack of efficacy [1]. Adaptive study designs are well established for cancer trials [2], and while they are applied to a lesser degree in other indications, a limited search in ClinTrials.gov revealed nearly 95 non-oncology industry sponsored adaptive trials over the past decade.
Regulatory Perspective on Adaptive Designs
As interest in flexible design elements grows across all indications, both the FDA and EMA have issued updated guidance on adaptive trial designs for drugs and biologics in recent years [3, 4]. For early phase exploratory studies, the FDA touts the advantages of an adaptive design that advises dosing, pharmacokinetic (PK) and pharmacodynamic (PD) responses, which may improve the design and possibly the chances of success of later-phase trials [3].
Adaptive Early Clinical Trial Designs
For single ascending dose (SAD) and multiple ascending dose (MAD) studies, adaptive design elements can be incorporated into the study protocol to add flexibility. This can be a valuable approach when there are still many unknowns in the initial clinical drug development phases. Case examples of recent adaptive protocols are highlighted in Table 1. These elements, such as alterations to drug dose, number of cohorts, or schedules of PK sampling and PD assessments, must be pre-specified in the protocol. For each adaptable component, limits should be set to address any safety concerns and to operate within acceptable risk parameters [5]. For example, if a blood sample schedule can be adjusted to optimize evaluable PK or PD data, the maximum number of blood draws should be noted in the protocol and not exceeded.
Table 1. Case Studies of Early Clinical Trial Adaptive Study Designs
Case Study
Study Design
Adaptive Elements
Implementation
Adaptive Dose and PK
2-part SAD and MAD study in healthy volunteers
Drug dose, infusion volume and/or rate, PK schedule (time points and number of samples)
Adaptive elements were modified based on AEs, clinical finding, safety and tolerability resultsProtocol allowed for reduced PK sample number if data permitted
Adaptive Cohort Number
SAD study in healthy volunteers
Cohort number and dose levels defined for groups 1-5 Option to add up to 2 additional cohorts
Upon review of cohort 1-5 safety and PK data, the decision will be taken to continue with increased dose levels, or an intermediate dose, or to cancel cohorts 6-7
AdaptivePD Response
MAD study in participants with obesity
Number of PD assessments
Multiple PD assessments evaluated in cohort 1 with option to omit or adjust PD schedule in subsequent cohorts Protocol also indicated the maximum number of PD assessments that can be taken
Adaptive Sample Size
Fixed-dose combination vs free dose cross-over bioequivalence (BE) study in healthy volunteers
Sample size re-estimation Clinical study activity paused during COVID-19 pandemic before all participants completed cross-over scheme
Adaptive and interim analysis was performed to re-evaluate study sample size and power, using a validated method for crossover BE design [6]. Trial results demonstrated BE based on completed participant data [7]
Advantages of Adaptive Early Clinical Trials
With pre-defined adaptive protocol elements, no amendments are required, which can save time and result in scheduling efficiencies. In the case of future pandemic outbreaks or other extraordinary situations, adaptive designs can serve as a mitigation step and potentially ‘save’ a study from timely delays or the need to repeat the trial. This approach was applied in a BE study, where sample size was re-assessed midway through trial conduct using validated statistical methods to complete the trial without further interruption from a COVID-19 outbreak [7]. Ultimately, the advantage of adaptive, early clinical trial designs can lead to both time and costs savings. Celerion’s Protocol Design and Development Scientists are trial experts and can design early clinical trials with flexible and adaptive elements to get the most out of a study, for less.
References
1. Pallmann P et al. BMC Med. 2018;16(1):29.
2. Bothwell LE et al. BMJ Open. 2018;8(2):e018320.
3. Food and Drug Administration. Adaptive Designs for Clinical Trials of Drugs and Biologics Guidance for Industry. 2019. https://www.fda.gov/media/78495/download.
5. Lorch U et al. BMC Med Res Methodol. 2014;14:84.
6. Potvin D et al. Pharm Stat. 2008;7(4):245-62.
7. Csonka D et al. Pharmacol Res Perspect. 2021;9(5):e00846.
Celerion continues its Celebration of Translating Science into Medicine for over a decade by highlighting how Celerion ScienceSM has contributed to the development of new therapeutics using our core competencies. While Celerion’s inventive spirit began over 50 years ago, our most important contribution to human health is happening right now, against COVID-19.
2020 has been a breakthrough year working with our biopharma clients to rapidly accelerate both vaccine development and therapeutic treatments against COVID-19. The safety of our patients and employees is an integral aspect of bringing all of us Closer to a Cure and Celerion is proud to have created one of the most comprehensive bioanalytical offerings in the industry for COVID-19 testing, with reliable and rapid results for SARS-CoV-2 PCR testing and antibodies against the virus.
Celerion remains at the forefront of innovative and novel technology to accelerate drug development, deliver high quality data and ensure participant safety. Over the past decade, Celerion has introduced several innovative, High-Tech systems.
Our Top 10 Innovative Technologies Implemented over the Past Decade:
2020 → Celerion delivers the Future of Pharmacy with state-of-the-art pharmacy suites for extemporaneous API compounding. The suites provide dedicated positive and negative pressure rooms for hazardous and non-hazardous material. In addition, the Lincoln pharmacy houses a devoted ADME suite and all Celerion pharmacies are USP <795>, <797>, and <800> compliant.
2020 → The latest enhancement to drug development is high-resolution mass spectrometry (HRMS). HRMS determines the exact mass of molecular ions and is applied in drug development to support in vitro metabolite identification as well as human ADME profiling studies. Our HRMS system is available at our Zurich bioanalytical laboratory.
2018 → FibroScan® is a noninvasive ultrasound-like device that simultaneously measures liver fat and fibrosis. FibroScan® is an integral part of inclusion and exclusion criteria for early phase nonalcoholic steatohepatitis (NASH) studies. Available at our Lincoln, NE and Phoenix, AZ clinics, we have a large database of FibroScan pre-screened participants.
2018 → Celerion provides a fully automated early clinical trial data management platform through the integration of Celerion’s proprietary electronic data acquisition system, ClinQuick®, with OmniComm’s TrialMaster® electronic data capture solution. This integration automates Celerion’s data acquisition system and provides consistent data management and reporting capabilities in one centralized database. It facilitates consistency of data collection across clinical sites, ensuring accurate and high-quality information.
2017 → Celerion uses biometric fingerprint technology to complement Verified Clinical Trials participant database registry. The registry enhances the quality and safety of clinical trials. Biometric fingerprint augments the accuracy and speed of verification as well as adding another layer of protection to ensure dual enrollment in a clinical trial does not occur.
2016 → Celexus® is Celerion’s client data information portal. Clients can access real-time clinical data with operational key performance indicators, a centralized repository for study documentation. The system also features a dynamic interactive experience for analyzing and interpreting clinical data.
2015 → Flow cytometry is a unique bioanalytical service offering. This technique is used to detect and quantify characteristics of a cell population or particles. Flow cytometry can measure T cell and NK cell panels as well as specific cell population isolations (CD cell molecules). Our system even determines simultaneous measurement of multiple cytokine, chemokine, immunoglobulin, or cell signaling targets from a single sample. Immune cell monitoring can be further investigated with ELISpot (enzyme-linked immunosorbent spot). ELISpot is useful to measure B-cell antigen-specific antibodies and T-cell secretion of IFN-γ. This bioanalytical service offering was introduced in 2017.
2014 → Lung clearance index is a sensitive measure of airway ventilation, able to evaluate early signals of efficacy for cystic fibrosis drug development. The system is available at Celerion’s Respiratory Center of Excellence in Belfast, UK, which also houses a dedicated on-site bronchoscopy suite allows bronchoalveolar lavage (BAL), whole-body plethysmography system as well as spirometry apparatuses.
2011 → Celerion’s highly automated ECG core lab uses an ECG acquisition Holter device to collect continuous digital 12-leads ECG recordings for cardiodynamic and safety ECGs. LCD screens optimize data quality by allowing visual inspection of all 12 leads before each collection time point. Digital recordings enable prompt onsite or remote review of safety ECGs to address potential adverse events or subject safety concerns. Using Bluetooth technology, this system was updated in 2015 for direct data capture.
2010 → Workload can be streamlined at the Speed of Science with Labnotes, an electronic bioanalytical laboratory notebook system that comprehensively captures study procedures, observations and results. The system ensures only reagents, solutions, equipment, and prepared standards within quality requirements are used (e.g. expiry, calibration, and preventative maintenance). Data can be reviewed immediately, reducing the chance of errors. Labnotes enables us to meet regulatory requirements for GxP, 21 CFR Part 11, and the FDA’s Electronic Records and Signature Rule.
by Sabina Paglialunga, PhD – Director, Scientific Affairs, Celerion
COVID-19 is a highly infectious respiratory disease caused by the SARS-CoV-2 virus that has affected every corner of the world and nearly all aspects of daily life.In a subset of COVID-19 patients, an exaggerated immune response can lead to acute respiratory distress syndrome (ARDS) requiring mechanical ventilation and leading to death. For society to return to “normal”, it is estimated that ~70% of herd immunity is required [1], which could result in thousands of casualties. Furthermore, this herd immunity target may not be achievable because it is still unknown if those with mild or asymptomatic cases of COVID-19 may not have built up sufficient immunity to prevent reinfection. Therefore, we must consider vaccination as the only viable option to eliminate this virus and thus the race is on to develop an effective vaccine against SARS-CoV-2.
Under the Microscope:
SARS-CoV-2 is a single stranded RNA virus, it is encapsulated by proteins and lipids. The virus has four main structural proteins; a spike glycoprotein (S), a small envelope glycoprotein (E), a membrane glycoprotein (M) and a nucleocapsid protein (N) in addition to other accessory proteins. A homotrimer of S-proteins facilitates binding to angiotensin-converting enzyme 2 (ACE2) receptor on host cells and cell entry [2]. The S-protein is therefore a key site for antibody neutralization, but vaccines developed against other viral protein are also under investigation.
Vaccines 101:
Vaccines boost immunity against infectious diseases through controlled exposure of an antigen, which can be an attenuated virus or fragments of viral proteins.The immune system responds by generating antibodies that protect against future infection. Subsequent exposure to the virus or another infected individual then triggers antibody recognition and the virus is cleared via the immune system activation.Adjuvants are applied to vaccine formulation to upregulate the antigenic response, and depending on the duration of protection, an additional booster shot may be needed.
Vaccine Safety Assessments:
Early clinical phase studies focus on safety, tolerability and immunogenicity, and throughout vaccine development particular attention is paid to hypersensitivity. While vaccines are generally considered safe, serious anaphylactic adverse events associated with immunization can occur, albeit they are extremely rare.Hypersensitivity to the antigen, adjuvants and preservatives have been observed [3] and may require dose adjustment or re-formulation. Another aspect to consider during vaccine development is the potential to induce a Th2 response. Th2 is one of two T-cell responses stimulated when antigens are presented to T cells. The type of T cell response (Th1 vs Th2) results in a particular set of cytokines released. Vaccines depend on a Th1 response to generate immunoglobulins, which elicit immunity against viruses, bacterial and fungal infections. A Th-2 response can counteract Th1 by upregulating interleukin-10 which has anti-inflammatory function [4]. If a Th2-type response is established upon immunization, it can prevent Th1-type response as these are antagonistic processes and a Th2-bias can potentially exacerbate the infection [5].
Bioanalytical Support of Vaccine Trials:
Advanced bioanalytical assays are needed for efficacy and safety measures. It is important that these bioanalytical tests are robust and analytically validated for their context of use in order to support a clinical trial [6].A variety of assays are available for vaccine drug development, which induce antibody titer, qPCR, ELISpot, cell profiling and cytokine biomarkers.
Table 1. Bioanalytical Assays for Vaccine Development
Bioanalytical Assays
Utility
Technology
Antibody titers
Determine the amount of antibodies produced in response to inoculation
Ligand binding methods such as ELISA or MSD
Neutralizing activity of the antibody
Determine the extend in which antibodies can clear the virus
Cell-based assays
Viral load
Determine the presences and amount of virus
qPCR
DNA vaccines
Quantify exogenous viral DNA components
qPCR
mRNA vaccines
Quantify exogenous viral mRNA components
qPCR
T-cell profiling
Examine immune cell populations
Flow cytometry
T-cell response
Examine IFN-γ activation
ELISpot
Antibody producing cells
Evaluate ex-vivo stimulation of PBMC for antibody production
ELISpot
Inflammatory cytokines
Monitor the ‘cytokine storm’ and potential risk stratify for disease severity
Ligand binding methods such as ELISA or MSD
Neutralizing antibodies (NAbs) assay
Antibodies that develop against the biotherapeutic product that can impact it efficacy or safety profile
Cell-based assays
Antibody Dependent Enhancement (ADE)
Characterize the causes of acute lung injury that may occur following coronavirus vaccination
Flow cytometry
Innovative Vaccine Platforms:
There are several platforms for vaccine development.Each technique holds unique advantages and challenges when it comes to safety, manufacturing and scalability [7-9]. The following explores these considerations for the various vaccines under development during the current pandemic.
Whole inactivated and live-attenuated virus vaccines: To create a whole inactivated vaccine, the virus is cultured in a laboratory and then killed (inactivated) or weakened (attenuated) with chemicals, heat, or radiation. This process conserves the virus structure, induces neutralizing antibodies and has been applied for other infectious diseases. There is a risk of hypersensitivity and Th2 bias. In the current crisis, a significant amount of live virus would need to be cultured quickly. Viruses are cultured in cell or egg based media, and these vaccines are contraindicated for individuals with an egg allergy. For recombinant live-attenuated vaccines, parts of the genetic sequence of the virus are manipulated to reduce the virulence. The antigens are produced in the body to facility an immune response. There is potential for the virus to revert to a virulent strain therefore this strategy may not be appropriate to inoculate sensitive populations.
Viral vector vaccines: Weakened adenoviruses or measles viruses are genetically engineered to produce SARS-CoV-2 surface proteins in a patient to elicit an immune response. There is a risk of anti-vector (adenovirus or measles) immunity, lowering the potential immune response against the SARS-CoV-2 target.
Subunit vaccines: Peptide components or fractions of the surface protein antigens are synthesized to create a vaccine.This strategy has a good safety profile however in most cases, subunit vaccines require adjuvants and booster doses.
Virus-like particles: The viral outer shell lacking the genetic material is introduced to patients to trigger a strong immune response. By conserving the virus structure, multiple antigens can be displayed. A caveat of this process is that manufacturing on a large scale may be technically challenging.
Nuclei acid vaccines: DNA or mRNA based vaccines use the patient’s own cell to generate virus peptides and surface proteins that will trigger an immune response (eg. S-protein). The mRNA is encased in a lipid layer that permeates the patients’ cells which act like a bioreactor and transcribe the mRNA into the pathogenic protein that will then stimulate an immune response. DNA vaccines work similarly to mRNA vaccines, with the antigen being coded in a DNA sequence. The DNA is translated to RNA then transcribed to the antigenic peptide. A DNA vaccine does require an extra step to enter into cells, typically with electroporation. One advantage is that more than one viral antigen may be coded. Nucleic acid vaccines are a relatively new technology and have not yet been approved for other infectious diseases.
Table 2. Benefits and Considerations for COVID-19 Vaccine Platforms
Vaccine Type
Benefits
Considerations
Whole inactivated virus
• Conserves viral structure
• Rapid development
• Potential for hyper-sensitivity and Th2-bias
• Culturing live virus
Live-attenuated recombinant virus
• Site-directed attenuation
• Potential to revert to virulent strain
• Not suitable for sensitive populations
• Increase the safety profile; non-integrating
• Egg and cell free
• Rapid, inexpensive and scalable manufacturing
• May suffer from instability
• Low immunogenicity
DNA vaccines
• Non-infectious
• Egg and cell free
• Greater stability
• Potential for multiple antigens
• Rapid production
• Specialized delivery system required
• Potential for integration into human genome
• Low immunogenicity
Adapted from: Zhang et al. 2019 [8] & Prompetchara et al. 2020 [9].
A Coordinated Effort:
The urgency of developing, validating and disseminating a COVID-19 vaccine is palpable. Nearly 100* sponsors have shifted resources and pivoted to the COVID-19 indication. Regulatory agencies are even cutting red-tape to expedite clinical trials. The MHRA approved a COVID-19 vaccine trial in 7 working days. In addition, regulatory authorities have also been swift to implement a series of guidance documents to support sponsors and CROs. Recently, the FDA issued guidance on development and licensure of vaccines to prevent COVID-19. In Europe, similar guidelines have been released by EMA and MHRA. Moreover, countries worldwide are ramping up production of syringes, vials and related paraphernalia needed to inoculate millions once a vaccines is approved. This level of swift global coordination has not been seen before.
Ending the Pandemic:
To end the COVID-19 health crisis quickly, we’ll need more than one solution.Biotech and pharma sectors as well as regulatory agencies and CRO stakeholders are up to the challenge as the race for a vaccine is well underway. Promising early results from vaccine developers such as the University of Oxford with a viral vector vaccine, and Moderna with an mRNA vaccine, are hopeful signs that relief is on its way.With a number of different types of vaccines under investigation, this increases our chances of developing several safe and effective COVID-19 vaccines.
2. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181(2):281-92 e6.
3. McNeil MM, DeStefano F. Vaccine-associated hypersensitivity. J Allergy Clin Immunol. 2018;141(2):463-72.
4. Berger A. Th1 and Th2 responses: what are they? BMJ. 2000;321(7258):424.
5. Rosenthal KS, Zimmerman DH. Vaccines: all things considered. Clin Vaccine Immunol. 2006;13(8):821-9.
6. Kar S, Islam R. Rapid and robust bioanalytical assays are critical for SARS-CoV-2 therapeutic and vaccine development and beyond. Bioanalysis. 2020.
7. Thanh Le T, Andreadakis Z, Kumar A, Gomez Roman R, Tollefsen S, Saville M et al. The COVID-19 vaccine development landscape. Nat Rev Drug Discov. 2020;19(5):305-6.
8. Zhang C, Maruggi G, Shan H, Li J. Advances in mRNA Vaccines for Infectious Diseases. Front Immunol. 2019;10:594.
9. Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38(1):1-9.
*GlobalData search on 01-June-2020. GlobalData, John Carpenter House, UK.
Acknowledgments: Thank you to Celerion scientists Aernout van Haarst, Sumit Kar, Michelle Combs and Lorraine Rusch for editorial assistance.
Biosimilars are not an exact copy but are similar to the originally approved biological product. Following an abbreviated pathway, they demonstrate equivalent PK, toxicity, similarity, and no clinical change compared to the innovator. The goal of a biosimilar is to introduce lower cost alternatives that can help improve patient access to biological treatments.
Celerion is the premier CRO for PK/PD assessments in healthy subjects and small patient groups. Building upon our bioequivalence and bioavailability expertise, we can design appropriate comparative studies to establish biosimilarity with marketed reference drugs that rely on pharmacodynamic biomarkers and potentially avoid larger patient studies.
With a legacy of over 50 years in clinical research, this year marks a decade of translating science to medicine as Celerion.To commemorate our 10-year anniversary, we are highlighting 10 years of Biosimilars experience.
Our Top 10 lists of Biosimilars Turn-Key Programs and Bioanalytical Assays:
Adalimumab: Monoclonal antibody that targets and inhibits tumor necrosis factor α (TNFα) activity. Adalimumab is indicated for rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, psoriasis, hidradenitis suppurativa, uveitis, and juvenile idiopathic arthritis. Marketed as Humira, the US patent expired in 2016.
Bevacrizumab: Monoclonal antibody that inhibits vascular endothelial growth factor A (VEGF-A). Bevacrizumab is indicated for colon cancer, lung cancer, glioblastoma, and renal-cell carcinoma and age-related macular degeneration.Marketed as Avastin, the patents expired in the US in 2019 and in Europe in January 2022.
Teriparatide:A recombinant 34 amino acid portion of human parathyroid hormone (PTH), indicated for osteoporosis. Originating product, Forteo/Forsteo patent expired in 2019.
(Peg)Filgrastrim: Neupogen is the originator filgrastrim product recombinant granulocyte colony-stimulating factor (G-CSF) indicated for conditions of neutropenia. Neulasta is a pegylated form of filgrastrim. Pegylation increases the half-life and stability. Neupogen patent expired in 2006 in Europe and 2013 is the US. Neulasta patent expired in 2015 and 2017 in the US and EU respectively.
Ustekinumab: Marketed as Stelara, it targets IL-12 and IL-23 and is indicated for plaque psoriasis, Chron’s disease and ulcerative colitis. Patents expires in 2023 in the US and in 2024 in Europe.
Omalizumab: Sold under the brand name Xolair, omalizumab is indicated for asthma and chronic idopathic utricaria. By inhibiting immunoglobulin E from binding to high affinity receptors on mast cells and basophils, omalizumab reduces sensitivity to allergens.Patent expired in 2017 in the US and Europe.
Ranibizumab: Ranibizumab is a monoclonal antibody fragment that inhibits angiogenesis by inhibiting VEGF-A. It treats age-related macular degeneration, a common source of vision loss with aging. It is also effective in diabetic macular edema. The brand name is known as Lucentis and its patent expires in June 2020 in the US and 2022 in Europe.
Cetuximab: A monoclonal antibody that inhibits epidermal growth factor receptor (EGFR) used for the treatment of metastatic colorectal cancer, metastatic non-small cell lung cancer and head and neck cancer. The patent for the brand name product, Erbitux, expired in 2014 in Europe and in 2016 in the US.
Trastuzumab: Monoclonal antibody, sold under the brand name Herceptin. Trastuzumab is a HER2 receptor antagonist, indicated for breast and stomach cancers that are HER2 receptor positive. Patents expired in 2014 in the EU and 2019 in the US.
Etanercept: Commonly known as Enbrel, it is a TNFα inhibitor that functions as decoy receptor for the cytokine. Etanercept is indicated for various rheumatic and psoriatic disorders. US patent is extended to 2028, however the patent is expired in EU and biosimilars are available in this region.
The global COVID-19 pandemic has altered the mechanics of nearly every industry. For early phase clinical trials, adapting to this crisis has led to augmented risk mitigation procedures and greater use of innovative technology. Since continuity of your drug program is important to you, the patients in need of these therapies, it is important to us.
We have taken a proactive approach to conducting business, now when research is needed more than ever. Our strengths lie in being flexible and nimble, which is a benefit during times of uncertainty.
Our ‘New Normal’ is built on three key pillars; Safety, Resources and Communication.
Safety First
The health and safety of our participant and staff is of the utmost importance. We have incorporated several measures to screen, clean and isolate for COVID-19 virus during trial operations. We are following all CDC, FDA and MHRA guidelines as well as local regulations for clinical operations during the COVID-19 pandemic.
Health Checks: All participants, staff and visitors will have their temperature checked and queried about COVID-19 symptoms or contact with positive patients upon entering Celerion facilities. Access will be denied to anyone showing symptoms of the disease.
PPE: All participants, visitors and staff will wear appropriate PPE while at the Clinic.
COVID-19 Testing: Regular PCR screening for virus detection and antibody testing for potential immunity will be performed.
Cleaning: Amplified cleaning measures have been put in place with special attention being paid to high-touch areas. Regular hand washing is encouraged for all participants and staff, and hand sanitizer is available at facility entry points and throughout the building.
Social Distancing: Isolation and distancing procedures are now in place. Participants will E-consent prior to screening and experience staggered screening as well as segregation in the clinic. Staff are instructed to work within their zoned areas, maintain 6 ft. social distance as much as possible, limit face-to-face interactions and time spent in common areas.
Testing, Testing, Testing! Celerion Bioanalytical Services has focused capabilities on COVID-19 screening by PCR, viral load quantification, antigen detection, antibody titer measurement, cell-mediated immunity assessments as well as cytokine and chemokine inflammation biomarker assays.
We Lead Quality
We have the essential resources in place to safely and effectively execute your clinical trial.
Supply Chain: We have secured the necessary PPE for our staff and participants.
Celerion Express:New screening facility located at the Nebraska Innovation Campus.
Bed Capacity and Space: 3 Clinical Pharmacology Units – 600 bed capacity
Enough beds to accommodate social distancing during participant confinement
Recruitment: Robust database of +130 000 participants
Experienced Workforce: +1000 Employees
Thoroughly trained Clinical Associates
Medical and Subject Matter Experts
Clinical Pharmacology team with expertise in study design, data analysis, interpretation and medical writing
Knowledgeable Bioanalytical Scientists
Communication is Key
With conditions rapidly changing, effective communication among all stakeholders is critical.
Response Plan: A Celerion Pandemic Management Plan is distributed to our clients prior to study start. This report outlines risk mitigation steps, training procedures and screening activities.
Adapting to Regulatory Guidance: We are in regular communication with IRB and IEC, and we are quickly disseminating and incorporating FDA, EMA, MHRA guidance regarding the COVID-19 pandemic.
Leveraging technology: We engage innovative tools to support virtual meetings and remote data review. Our clients are provided with real time data accesses through our Celexus® system.
Celexus® is Celerion’s client data portal. It is an information hub with real-time access to safety data and trial progress dashboard.
During this dynamic situation, the steps we have put in place for the safety of participants and staff, securing supply chain and innovative technology are designed with one objective in mind, to help our biotech and pharma partners get their products to market faster.
by Sabina Paglialunga, PhD – Director, Scientific Affairs, Celerion
SARS-CoV-2 symptoms include fever, cough and shortness of breath. The disease is especially hard on vulnerable populations such as the elderly and those with underlying health conditions like obesity and hypertension. Now, developing reports indicate that children may also be at great risk than originally thought.
Into the Eye of the Storm
The virus primarily enters the body via the respiratory tract, and into lung cells through ACE2 receptors. To counteract, the body mounts an immune response to defend against the foreign virus. However, this attack tends to occur several days after initial infection, by when significant amount of viral shedding has already occurred, contributing to the wide spread of the disease. As part of the defense process, there is an influx of immune cells, which include macrophages and T cells to the respiratory tract, leading to an overall systemic upregulation of pro-inflammatory cytokines (e.g. IL-6, IL-1β, TNFα) as well as interferon (IFN) signaling and production anti-viral factors. The resulting immune response drives B cells to create antibodies against SARS-CoV-2. These antibodies recognize the virus, neutralize and clear it from the body.
While many infected individuals are asymptomatic or present with mild symptoms. Since the ACE2 receptors are also expressed in gastrointestinal tissues, abdominal symptoms are associated with COVID-19. More severe cases will result in pneumonia, sepsis, acute respiratory distress syndrome (ARDS) requiring hospitalization and even death. Researchers have found that a hyper-immune response is associated with the most severe COVID-19 cases. Early studies from China reported patients in intensive care units (ICU) had dramatically elevated levels of IL-6, IL-10 and TNFα, accompanied by a reduction in CD4+ and CD8+ T cells [1]. This hyper-, pro-inflammatory state is often referred to as the cytokine storm or cytokine release syndrome (CRS). In this state, pro-inflammatory cytokines are released in excess amounts, creating an exaggerated cytokine response. This can lead to not only damage to the virus and infected host cell but also healthy cells too. In the lungs, destruction of areole cells can result in hypoxia and ARDS, which may lead to death. Similar finding of increased levels in IL-2, IL-7, G-CSF, CXCL10, MCP-1, MIP-1α, and CXCL10, CCL7 were subsequently reported, suggesting the hyper-inflammation associated with severe COVID-19 exacerbates lung damage [2, 3]. The cytokine storm is thought to be driven by IL-6 [4]. In that respect, the virus also infects monocytes, macrophages and dendritic cells that can result in their activation and further secretion of IL-6.
Navigating the Current Pandemic with Past Coronavirus Experience
The raging cytokine storm also contributed to morbidity in patients infected with other Coronaviruses. There are seven known human coronaviruses. The 229E, HKU1, NL63, OC43 strains are associated with the common cold, however more virulent forms have resulted in major past outbreaks; SARS (Severe Acute Respiratory Syndrome) in 2002 and MERS (Middle Eastern Respiratory Syndrome) in 2012. SARS, MERS and COVID-19 viruses originated in bats then transitioned in an intermediate host before jumping to humans (reviewed in [5, 6]). The SARS-CoV-2 virus shares similar sequence identity to the SARS virus (SARS-CoV) and may provide insight into the current pandemic and cytokine storm.
SARS-CoV-2 binds ACE2 receptors more efficiently than the SARS-CoV 2003 strain but less efficiently than the strain first identified in 2002, as mutations in SARS-CoV created a less virulent form. It is too early to tell how or if any mutations in SARS-CoV-2 will affect infection and mortality rates. The reproductive number (R0), number of cases directly generated by one infected individual, as well as complication and mortality rates for SARS, MERS and COVID-19 is listed in Table 1. While the infection rate of COVID-19 is much greater than SARS and MERS, the mortality rate is expected to be lower. On the other hand, ARDS-specific complication of COVID-19 is anticipated to be similar to SARS and MERS.
Table 1. Virulence of SARS, MERS & COVID-19
Characteristics
SARS
MERS
COVID-19*
Reproductive number (R0)
1.7 – 1.9
<1.0
2.0 – 2.5
ICU admission (% cases)
23 – 24%
53 – 89%
24%
ARDS-specific complication (% cases)
20%
20 – 30%
18 – 30%
Mortality rate
14 – 15%
35%
2 – 4%
Adapted from [5-7]. *Current estimated values.
A key lesson learned from previous outbreaks is how Coronaviruses commandeer the host cells immune response unleashing the cytokine storm. Both SARS-CoV and MERS-CoV encode accessory proteins known to antagonize IFN and suppress its signaling [8]. Diminished IFN response, allows for unchecked and rapid viral replication in which a surge of cytokines and chemokines are eventually and robustly released in a last ditch attempt to kill the virus contributing to the cytokine storm. Moreover, this dampening of the IFN response is strongly associated with disease severity [9]. In a second strike, the SARS-CoV virus also encoded accessory proteins that activate NLRP3 inflammasome, which drives IL-1β and promotes expression of other pro-inflammatory cytokines such as TNFα and IL-6 (reviewed in [6]). These strategic viral battle maneuvers are postulated to contribute to the cytokine storm that is being observed with the current COVID-19 pandemic.
The Next Wave: Kawasaki Disease-like Symptoms in Children
While early reports suggested that children were spared from COVID-19, a number of significant severe COVID-19 cases in pediatric populations have emerged [10, 11]. More urgently, is the rise of multisystem inflammatory syndrome in children (MIS-C) clusters arising in the US and EU associated with COVID-19. As of May 12, 2020 there were over 50 cases reported in New York City alone. With the current situation escalating rapidly, on May 14, 2020 the CDC issued a health advisory for MIS-C. The disorder is associated with active infection or antibodies against SARS-CoV-2 [12], or COVID-19 exposure within 4 weeks prior to the onset of symptoms. Clinically, MIS-C is defined as persistent fever, requiring hospitalization with multisystem involvement (cardiac, renal, respiratory, hematologic, gastrointestinal, dermatologic or neurological) and Kawasaki disease-like symptoms.
Kawasaki disease is a rare, vasculitis disorder affecting young children that can lead to coronary artery aneurysm, thrombosis and sudden death if left unchecked. It is the leading cause of acquired heart disease in pediatric populations in developed countries. Disease diagnosis is established by presence of persistent fever along with four of five signs and symptoms; bilateral conjunctivitis, strawberry tongue and/or dried cracked lips, polymorphous exanthema, cervical lymphadenopathy, swelling of hands and feet. While the exact etiology is still not fully understood, it is thought that Kawasaki disease may arise from an infectious trigger (bacterial or viral), resulting in upregulation of the immune system in a genetically susceptible individual (reviewed in [13]).
Kawasaki disease has been previously reported with acute viral infection of the respiratory system, resulting in dysregulated immune response as observed in with the cytokine storm [13]. A children’s hospital retrospective review of Kawasaki disease admission from 2009 to 2013 linked the disease with rhinovirus and parainfluenza and to a lesser extent human coronaviruses strains 229E, NL63, OC43 [14]. Therefore, it is conceivable that SARS-CoV-2 may represent the infectious trigger during the current crisis. Similar to the cytokine storm described above, Kawasaki disease patients also have remarkably increased CRP, IL-6, IL-10 and IFNγ [13], and potential involvement of an acute B cell response [15]. Indeed, the first case of Kawasaki disease with concurrent COVID-19 infection was reported in a 6-month old infant with elevated CRP. The patient also had a continuous fever, lack of appetite, mild congestion, and met the classic Kawasaki disease criteria [16]. In addition, RT-PCR testing confirmed positive result for COVID-19 infection.
While the incidence of past Kawasaki disease cases tended to be seasonal and were more prevalent in Asian countries and Asian races in the US [13]. Genome-wide association studies have identified candidate loci [17], however the genetic factors contributing to pathogenesis of the disease are not fully understood. Moreover, it is still too early to determine if genetic susceptibility plays a role in the rash MIS-C observed in the current COVID-19 pandemic as children of many ethnicities have reported disease symptoms [12, 16].
Quieting the Storm: Immunosuppressant Drugs
Since disease severity appears to be driven by a cytokine surge, it suggests that anti-inflammatory treatments may quell the cytokine storm and improve disease prognosis. During the SARS outbreak, immunosuppressants such as corticosteroids resulted in more harm than good and are therefore not recommended for the treatment of COVID-19 [18]. For that reason, clinicians are expanding their toolbox and looking to therapies targeting pro-inflammatory cytokine. Tocilizumab, a marketed IL-6 receptor antagonist, is indicated for CSR, and was recently reported to improve COVID-19 patient outcomes in a small open-labelstudy in China. Further randomized controlled trials are now underway with Tocilizumab, as well as with other direct IL-6 antagonists such as Siltuximab and Sarilumab and indirect inhibitors such as Ruxolitinib. Additional immunosuppressant drugs such as anti-IL-1 anti-IL-17, anti-TNFα are also currently being explored as potential treatment under the compassionate use authority or off-label, with randomized clinical trials starting soon. A recent GlobalData* database search retrieved nearly 75 COVID-19 planned or ongoing trials with interleukin inhibitors. Moreover, there may be a role for novel anti-inflammatory drugs in COVID-19 treatment. Across all stages of development, sponsors are promptly advancing their program to help the fight. In response and support of this effort, the FDA issued guidancefor industry on developing treatment and preventative COVID-19 drugs.While, immunosuppressant drugs will not be a cure for COVID-19, only a vaccine or anti-viral drug can annihilate the virus, anti-inflammatory therapies can play an important role in reducing the need for ventilators and preventing mortality.
For children presenting with classic Kawasaki disease criteria, treatment guidelines include intravenous immunoglobulin and high dose aspirin [16]. Refectory Kawasaki disease may be treated with corticosteroids, although this approach remains controversial due to the risks associated with steroids [13]. Alternative therapies aiming to suppress the pro-inflammatory state include TNFα inhibitors such as Infliximab, Abciximab and Etanercept. Overall, treatment with these anti-inflammatory drugs led to improved outcome, reduction in fever and reduced risk of coronary artery aneurysms (reviewed in [13]. It remains to be seen how TNFα inhibitors will be deployed in the current pandemic.
Biomarkers beaming like a Lighthouse
Another area under intense investigation is the use of biomarkers. Cardiac biomarkers such as N-terminal pro-B-type natriuretic peptide (NT-proBNP) indicate cardiac stress can be useful to assess cardiac risk associated with Kawasaki disease. Furthermore,identifying early prognostic biomarkers of COVID-19 severity is important for clinicians to initiate therapy before critical care is needed. Low lymphocyte count as well as the serum levels of D-dimers, ferritin, CRP and IL-6 may help stratify mild from severe cases [19, 20]. C-reactive protein (CRP), an acute-phase inflammatory protein synthesized by IL-6-dependent signaling and indicator of IL-6 bioactivity. CRP can be used to predict the cytokine storm severity and monitor IL-6 blockade efficacy [21]. Therefore, monitoring chemokines and inflammatory cytokines may not only be helpful to risk stratify for disease severity but also demonstrate target engagement and proof-of-mechanism for immunosuppressant and anti-inflammatory drugs in development.
A Fleet of Bioanalytical Platforms
In addition to measuring cytokines in serum, COVID-19 testing will help launch us out of social isolation. A SARS-CoV-2 qPCR test can confirm the presence of the virus and antibody testing can determine if someone was already exposed and may have immunity to SARS-CoV-2. For clinical trials, it is important that these assays meet stringent validation criteria as well as demonstrate good sensitivity and specificity.
Utilizing an advanced array of bioanalytical tools, Celerion provides full bioanalytical solutions for small and large molecule assays as well as genetic and cell-based assays. For the health and safety of our participants and staff, we provide COVID-19 testing at our Clinical Pharmacology Units in Lincoln, NE, Phoenix, AZ and Belfast, UK. These tests are validated and performed in our very own Bioanalytical laboratory. Critical to the current pandemic, we offer analytically validated cytokine and chemokine assays utilizing a sophisticated automated electrochemiluminescence (ECL) platform.
As a full-service CRO and leader in early phase drug development, Celerion is ready to serve our biotech and pharma partners develop life-saving treatments during these extraordinary times. Our mission is to focus every day on helping our clients get their drugs to market, so that they touch the lives of our family, friends and people in need around the world.
References
Pedersen SF, Ho YC. SARS-CoV-2: a storm is raging. J Clin Invest. 2020.
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033-4.
Yang YS, C.; Li, J.; et al. . Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. medRxiv. 2020.
Moore BJB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020.
Petrosillo N, Viceconte G, Ergonul O, Ippolito G, Petersen E. COVID-19, SARS and MERS: are they closely related? Clin Microbiol Infect. 2020.
Fung SY, Yuen KS, Ye ZW, Chan CP, Jin DY. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect. 2020;9(1):558-70.
Ruan S. Likelihood of survival of coronavirus disease 2019. Lancet Infect Dis. 2020.
Wong LY, Lui PY, Jin DY. A molecular arms race between host innate antiviral response and emerging human coronaviruses. Virol Sin. 2016;31(1):12-23.
Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38(1):1-9.
Shekerdemian LS, Mahmood NR, Wolfe KK, Riggs BJ, Ross CE, McKiernan CA et al. Characteristics and Outcomes of Children With Coronavirus Disease 2019 (COVID-19) Infection Admitted to US and Canadian Pediatric Intensive Care Units. JAMA Pediatr. 2020.
Mehta NS, Mytton OT, Mullins EWS, Fowler TA, Falconer CL, Murphy OB et al. SARS-CoV-2 (COVID-19): What do we know about children? A systematic review. Clin Infect Dis. 2020.
Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020.
Turnier JL, Anderson MS, Heizer HR, Jone PN, Glode MP, Dominguez SR. Concurrent Respiratory Viruses and Kawasaki Disease. Pediatrics. 2015;136(3):e609-14.
Lindquist ME, Hicar MD. B Cells and Antibodies in Kawasaki Disease. Int J Mol Sci. 2019;20(8).
Jones VG, Mills M, Suarez D, Hogan CA, Yeh D, Bradley Segal J et al. COVID-19 and Kawasaki Disease: Novel Virus and Novel Case. Hosp Pediatr. 2020.
Elakabawi K, Lin J, Jiao F, Guo N, Yuan Z. Kawasaki Disease: Global Burden and Genetic Background. Cardiol Res. 2020;11(1):9-14.
Russell CD, Millar JE, Baillie JK. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet. 2020;395(10223):473-5.
Velavan TP, Meyer CG. Mild versus severe COVID-19: laboratory markers. Int J Infect Dis. 2020.
Henry BM, de Oliveira MHS, Benoit S, Plebani M, Lippi G. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): a meta-analysis. Clin Chem Lab Med. 2020.
Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun. 2020:102452.
*GlobalData search: Infectious disease therapy area; Coronavirus disease 2019 (COVID-19) indication; planned, ongoing not recruiting, ongoing recruiting, ongoing recruiting by invitation trial status; L04AC Interleukin inhibitors ATC classification. Search date 01May2020. GlobalData, John Carpenter House, UK.
Acknowledgments
Thank you to Celerion scientists Aernout van Haarst, Sumit Kar, Fred Pritchard, Michelle Combs and Lorraine Rusch for editorial assistance.
By Sumit Kar, Lead Scientist – Biomarkers, Celerion
Cytokine biomarkers are released in the body after SARS-CoV-2 viral particles are presented by antigen presenting cells initiating a cytokine storm.1 Cytokine storm, known clinically as haemophagocytic lymphohistiocytosis or cytokine release syndrome, is mostly seen after viral infections, and leads to constant fever, increased ferritin, and multi-organ failure including acute respiratory distress syndrome (ARDS). For this reason, anti-inflammatory drugs, such as some targeting IL-6, are being tested as therapies for COVID-19 to prevent ARDS, the most common reason for fatality in patients suffering from COVID-19.
SARS-CoV-2 studies show alterations in serum IL-2, IL-6, IL-7, granulocyte-colony stimulating factor, IP-10, MCP-1, MIP1-α, and TNF-α, which are positively correlated with COVID-19 disease severity.1 The exact cytokine profile varies between studies. In previous viral challenge trials with neutralizing antibody therapies (e.g. for influenza), only IP-10 and IFN-g were reduced after drug dosing.2 The specific cytokines needed for SARS-COV-2 trials are yet to be well-characterized. Therefore, larger multiplex panels measuring several markers together are recommended for speed – especially those that are well characterized for reliability (e.g. Meso Scale Discovery® (MSD) V-Plex Assays).
Use Cases of Cytokines for COVID-19 Clinical Trials
These cytokine biomarkers can be monitored in SARS-COV-2 trials for vaccines, antivirals, and antibody therapies. Cytokines can be measured for patient enrollment, mechanism of action, and treatment effect contexts of use. For example, drugs trying to prevent ARDS should measure cytokines as a secondary endpoint and to show their mechanism of action. At Celerion, we validate all biomarkers fit-for-purpose based on their context of use in the study following the latest regulatory guidelines.
Celerion’s Cytokine Assays
At Celerion, we have validated MSD Cytokine Panels with up to 10 cytokines and chemokines for quantification via automated electrochemiluminescence (ECL). The panels can be custom designed according to need while maintaining the performance of the assay.
Respiratory specific matrices (i.e., bronchoalveolar lavage fluid (BALF), sputum and saliva) can be a reservoir of cytokine upregulation upon viral exposure via inhalation.3 Celerion has extensive experience measuring biomarkers in these respiratory matrices using the latest technology, which quantitates cytokines at ultra-low concentrations via Quanterix® Simoa and MSD S-Plex platforms.
References
1. Mehta P, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. The Lancet. 396(10229), 2020
2. McBride JM, Lim JJ, Burgess T, Deng R, Derby MA, Maia M, Horn P, Siddiqui O, Sheinson D, Chen-Harris H, Newton EM, Fillos D, Nazzal D, Rosenberger CM, Ohlson MB, Lambkin-Williams R, Fathi H, Harris JM, Tavel JA. Phase 2 randomized trial of the safety and efficacy of MHAA4549A, a broadly neutralizing monoclonal antibody, in a human influenza a virus challenge model. Antimicrob Agents Chemother 61:e01154-17, 2017
3. Horiuchi T, et al. Biomarker profiles of BALF in ALI/ARDS due to pandemic (H1N1) 2009 influenza. European Respiratory Journal. 38 (Suppl 55), 2017
Acknowledgements
Special thank you to Celerion scientists Curtis Sheldon, Amanda Daugherty, and Aernout van Haarst for their editorial assistance.
In the therapeutic toolbox, there is a multitude of drug categories. For decades, small molecules have been the cornerstone of pharmacology. In recent years, biological products have ushered in a wave of therapies, and forged the path for other cell- and genetic- based treatments. Greater understanding of the microbiome in association with dysbiosis and associated diseases have led to the advancement of pharmabiotics.
How Microbes can Treat Diseases
Pharmabiotics or live biotherapeutic products (LBP) refers to live microbes administered to patients to treat a disease. Gut dysbiosis is an imbalance of digestive track bacteria species that may contribute to a host of diseases including depression, Alzheimer’s disease, diabetes, irritable bowel disease (IBD) and nonalcoholic fatty liver disease (NAFLD) and even cancer. Replenishing the so-called “good” bacteria may return balance and revert or alleviate symptoms of the conditions. Gut-brain-, gut-lung- and gut-liver- axes research has expanded our understanding of the microbiome and fostered a new branch of drug development. Dysbiosis has also been described in other organ systems such as the skin, mouth, vagina and placenta [1], all which may benefit from LBP treatment.
Pharmabiotics are validated through clinical trials with safety and efficacy endpoints for a given indication and follow the same regulatory pathway to approval as other novel drugs. In that respect, pharmabiotics differ from prebiotic and probiotic products. Prebiotics are fermented fibers that supports the growth of certain gut bacteria, while probiotics are live microorganisms that support or show potential for health benefits. For the most part, these two products are considered a dietary supplement or a medical food and do not require FDA premarket approval.
LBP can be derived from a human host or genetically modified or engineered to express a specific trait. According to ClinTrials.gov several LBP are in development [2]. This includes pharmabiotics for cancer adjuvant therapy, asthma, IBD and prevention from recurrent infection such as Clostridium difficile and bacterial vaginosis.
FDA Guidance on Pharmabiotics
The FDA defines LBP as a product that [3]: i) contains live organisms, such as bacteria; ii) is applicable to the prevention, treatment, or cure of a disease or condition of human beings; and iii) is not a vaccine.
The FDA guidance, which was updated in 2016, outlines requirements for describing the LPB and the manufacturing process as well as adjuvant substances if necessary. For non-clinical studies, the FDA recommends pharmacological and toxicological studies of the LBP in laboratory animals, or in vitro, to support a proposed clinical trial evaluating the investigational LBP. Similar to small molecule or biological drug development, non-clinical studies for LBP may include general toxicity; target organs or systems of toxicity; teratogenic, carcinogenic, or mutagenic potential of any ingredient in the product; and relationship of dosage and duration to toxic response and pharmacological activity. In addition, during early clinical Phase I studies, emphasis should be placed on subject safety. Early studies with healthy volunteers can be important to identify common LBP-associated adverse events before proceeding to studies in more vulnerable populations, such as those with the disease of interest. Currently, there is no EMA guidance for pharmabiotics as the agency does not regard LBP as a drug; however this stance may change as the field continues to grow.
LPB Dosing and Monitoring
Commonly, dosing units for LPB are based on a colony forming unit (CFU). According to the FDA, CFU is the measurement of viable microbial cells that are capable of replicating on agar plates and forming colonies which are then counted [3]. Depending on the targeted tissue, delivery of LBP may be orally ingested, administered via the urogenital track or other methods.
While traditional PK assessments may not apply to the field of pharmabiotics; microbiota diversity, taxonomic composition as well as the microbiome are important pharmacological endpoints. LPB bacterial strain colonization in stool and fecal metabolomic profile, including short chain fatty acids and bile acids, may be evaluated for LBP targeting gut disorders, while sputum may be obtained for microbe count and bacterial DNA for an asthma indication. Supporting serum biomarkers such as inflammatory cytokines, chemokines, and hormones can also demonstrate systemic pharmacodynamic changes.
Safety Considerations
While probiotics are generally considered safe for consumption, there has been reported instances of adverse events which can include systemic infections, deleterious metabolic activities, excessive immune stimulation in susceptible individuals and gene transfer [4]. Pharmabiotics may results in similar adverse effects, therefore close monitoring may be warranted. An excellent recent invited review by LeBegue et al. describes the history and study design considerations for pharmabiotic products [5].
The Celerion Advantage
Celerion clinics have experience with LPB and the unique challenges in sampling and handling key matrices such as feces, urine, sputum and other fluids for pharmabiotic studies. Our team of highly skilled Regulatory and Drug Development Service associates can support your LBP from IND through Phase II. In addition, we have a vast database of healthy volunteers as well as access to patient populations such as asthma, NAFLD and irritable bowel disease patients.
References
Belizario JE, Napolitano M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol. 2015;6:1050.
Early Clinical Trials With Live Biotherapeutic Products: Chemistry, Manufacturing, and Control Information; Guidance for Industry. . Food and Drug Administration; 2016.
Doron S, Snydman DR. Risk and safety of probiotics. Clin Infect Dis. 2015;60 Suppl 2:S129-34.
LeBegue CE, Love BL, Wyatt MD. Microbes as Drugs: The Potential of Pharmabiotics. Pharmacotherapy. 2020;40(2):102-6.
COVID-19 Vaccines in Development
COVID-19 is a respiratory illness caused by a novel coronavirus. According to the CDC, symptoms may include fever, coughing and shortness of breath. The virus is highly contagious and can spread between people who are in close contact with one another (within 6 feet) or when an infected person coughs or sneezes spraying respiratory droplets.
While the world is grinding to a halt to slow the spread of the COVID-19 through social-distancing, self-isolation and quarantine efforts. The race to find a COVID-19 vaccine has just began. As of March 18th 2020, there are 298 clinical trials for COVID-19 and 129 drugs in the pipeline which may potentially treat this disease (GlobalData).
As we face this ever-growing pandemic, Celerion is ready to work with our pharma and biotech partners to ensure safety and efficacy of COVID-19 vaccine development. Celerion is a leader in vaccine development with infections disease experience. Our track record spans both preventive and therapeutic vaccines, subunit and conjugates across all phases I-IV in more than 20 countries, 300 study centers and 4500 subjects.
We will get through this, together!!
Biological therapies such as peptides, enzymes and antibodies have changed the face of medicine, providing crucial treatment for a number of devastating inflammatory, endocrine and oncological diseases. However, these drugs often come with a hefty price tag due to the complicated nature of manufacturing biological products which are derived from living systems. In an effort to reduce drug prices, especially among biologicals, the Biologics Price Competition and Innovation Act was passed in 2010, creating a regulatory framework for biosimilar drugs to advance on to the market.
Biosimilars are not an exact copy but are similar to the originally approved biological product. While innovator products must demonstrate pharmacokinetic (PK), dose finding, efficacy, safety and clinical benefit/risk for approval; a biosimilar product follows an abbreviated pathway and must demonstrate equivalent PK, toxicity, similarity, and no clinical change compared to the innovator. With this framework, the goal is to introduce cost-effective alternatives to innovator biological drugs that can help improve patient access to these treatments.
The Biosimilar Model:
Robust, vigorous analytical characterization, justification of cell lines, examination of post-translation products, and preclinical studies are emphasized in the biosimilar paradigm. Most biosimilar programs only require one Phase I or III study, reducing cohort sizes and study time from traditional drug approval processes. Immunogenicity data must be collected in patients, however this can be descriptive in nature and is not required to be powered.
In lieu of a comparative efficacy study, pharmacodynamic (PD) biomarkers related to the mechanism of action can be evaluated as a study outcome. For example, picture a biosimilar product like recombinant insulin binding to a cell receptor to initiate a chain of signaling events. PD biomarkers such as glucose concentrations from these early events of the signaling cascade tend to be more sensitive, while late PD biomarkers like HbA1c are related to clinical outcome yet tend to have lower sensitivity. Therefore, early PD biomarkers are recommended as they are apt to illustrate how a drug behaves and can be applied to simulate PK/PD dose response. Both PK and PD biomarkers are required to be within margins to show equivalency, for the FDA these margins are 80-125%. When no good PD biomarker is available for an indication, clinical endpoints should be the same as the innovator product.
The FDA’s Action Plan:
While the EU has benefited from biosimilars for close to 15 years, with 45 approved products and marked reductions in drug prices [1], the US has been slow to embrace this technology. To date, 19 biosimilars have been approved in the US, however 7 products are still not on the market mainly due to patent litigation [1]. In an effort to cut-red tape, increase drug competition and reduce prices for patients, the FDA released their Biosimilar Action Plan in June 2018. This plan outlines their four-pronged approach to expedite biosimilar product development through; increasing efficiency in the approval process, clarifying regulatory guidance, providing educational support for patients and prescribers, and supporting market competition. Progress towards these goals is evident through 60 biosimilar development programs, a newly released final guidance for interchangeability, and plans to license biosimilar and interchangeable insulin products.
Optimizing Biosimilar Product Development:
The following study design recommendations are aimed to optimize biosimilar programs when planning for multiple authorities or indications, or for interchangeability designation.
Multiple Jurisdictions – Consider a 3-way crossover study with US and EU innovator and reference product to establish bridging for various markets.
Extrapolation – Consider running the study in the most homogenous patient group, this will improve sensitivity. Once approved for one indication, approval can be extrapolated to all other approved indications with justification based on totality of evidence.
Interchangeability– Refers to pharmacists switching between products without the consent of the prescriber. In the EU, interchangeability of a product is dictated by member states. Elsewhere, Australia has recently granted interchangeability designation for adalimumab biosimilar products [2] . In the US, interchangeability of a biosimilar should be demonstrated in a randomized two-arm (switching vs non-switching) clinical study with a reference product lead-in period. In a crossover design, the switching arm should have at least 2 or more product switches. One major challenge with interchangeability study design is that many antibody biosimilars have long half-lives and which would require a long cross-over study. However, the added statistical power of the cross-over design may well be worth the long study duration. In addition, during such trials, it is important to closely monitor for safety and adverse immune responses. Alternative approach is an integrated study design to demonstrate no clinically meaningful difference between reference and the biosimilar product and evaluate the impact of switching.
Turn-key Solutions for Biosimilar Programs:
As a global leader in analytical and clinical studies for biological drug development, Celerion provides turn-key solutions for a number of biosimilar programs.
• Adalimumab – Anti-TNFα biological for the treatment of arthritis, plaque psoriasis, ankylosing spondylitis, Crohn’s disease, and ulcerative colitis
• Rituximab – CD20 antagonist for autoimmune diseases
• Teripartide – Recombinant human parathyroid hormone for osteoporosis
• Ustekinumba– IL-12/23 inhibitors for psoriasis
• See our full list of validated bioanalytical assays at www.celerion.com/assays
Conclusion:
Altogether, biosimilars are primed to make a significant impact on access to vital medicines worldwide. Currently, of the marketed biosimilar products available in the US the cost for these products is an estimated 17-57% less than the originator price. With new, efficient tools to support biosimilar development, the FDA aims to see even more meaningful impact on drug prices.
Chronic obstructive pulmonary disease (COPD) describes a cluster of diseases linked to breathing problems and airflow blockage, such as emphysema and chronic bronchitis. COPD is often associated with cigarette smoking, and prolonged exposure to poor air quality or toxic gaseous pollutants. This chronic disease affects over 3 million people in the US each year, and is currently the third leading cause of death worldwide. COPD results in difficulty breathing due to limited airflow availability in the lungs, and symptoms include shortness of breath, wheezing or chronic coughing. Periods of sustained or severe COPD episodes are referred to as exacerbations. While there is no cure for COPD, current treatments increase bronchodilation (opening of the airways) to provide symptom relief. For nearly 50 years, bronchodilators such as beta-2-adrenoreceptor agonists and muscarinic antagonists have been at the cornerstone of COPD treatments and are available in; short-acting, long-acting, single-, dual- or glucocorticoids combined triple-therapy.
Over the last decade, a surge in COPD research has greatly expanded our understanding of the disease and the key inflammatory players involved in airway blockage. This global initiative has led to the clinical development of over 25 novel drug targets. Roflumilast, a phosphodiesteratse type 4 (PDE4) inhibitor, was the first approved COPD add-on therapy which specifically targets the inflammatory processes underlying COPD. Ongoing research has identified pivotal roles for neutrophils and eosinophils (inflammatory cells) in COPD development, and resulted in a number of exciting drug targets in the pipeline. By addressing the underlying mechanisms responsible for disease development, this may lead to treatments that alter the course of disease progression and possibly a cure for COPD.
Along with new drug targets, the COPD biomarker landscape has also changed. While spirometer and patient-reported outcome remain critical clinical study endpoints, a role for soluble biomarkers to characterize patient populations and demonstrate drug efficacy has emerged. Fibrinogen is a soluble biomarker drug development tool approved by the FDA for COPD patient selection. Plasma fibrinogen levels are elevated in patients with COPD and are likely to experience an exacerbation, a key inclusion criteria for clinical trials aiming to demonstrate a reduction in exacerbation rates. In addition, validated assays for pro-inflammatory cytokines such as TNFa, IL-5, IL-8 and IL-17 are also of interest as increased levels of such cytokines may reflect an upregulation of neutrophilic and eosinophilic immune cell activity, and their attenuation can be indicative of reduced inflammation. Moreover, these biomarkers can be measured in either serum or right at the site of the airway blockage and inflammation, in lung fluid. Sputum collection (coughed up saliva and mucus mixture) and bronchoalveolar lavage (BAL) are two manners to retrieve lung fluid secretions. BAL is a minimally invasive endoscopic technique performed by a trained bronchoscopist to obtain cellular and biochemical components from lung fluid during a saline wash. Various cell types, cytokines and drug concentrations can all be measured to better understand pharmacokinetic – pharmacodynamic relationships.
With the development of new technologies and more sensitive bioanalytical assays, novel, non-invasive breath tests have entered the investigational scene in recent years. For instance, methodologies have been applied in exploratory clinical studies to analyze volatile organic compounds (VOC) in breath, exhaled breath condensate (EBC), and particles in exhaled air (PExA). Due to the nature and origin of VOCs which are derived from the entire body and microbiome, metabolomic analyses of VOCs have been explored as a potential tool to support early diagnosis of a broad range of systemic diseases, but it may also be useful for respiratory disease. For instance, VOC biomarkers have been shown to correlate with sputum markers from inflammatory cells and cell counts in COPD. Distinct patterns have also been associated with COPD disease staging. In contrast to VOC, EBC and PExA analytes are considered respiratory tract-specific, reflecting airway lining fluid and immune cell mediators from the lower airways. Their analysis allows identification and quantification of inorganic anions and cations, proteins, lipids and genes known to play a role in immune response. In addition, drug concentrations can be assessed in EBC and PExA samples.
Altogether, novel biomarkers and non-invasive breath test technologies may help diagnose respiratory pathologies, identify pathogens and distinguish treatable traits. Moreover, these innovations in COPD may provide new insights into inflammatory pathways in relation to pulmonary disease and disease stages. Finally, novel biomarkers in exhaled breath are likely to provide new tools to monitor disease state and treatment effects for specific drug targets.
Nonalcoholic steatohepatitis (NASH) is a chronic liver disease that affects over 17 million Americans and this number is growing. NASH can lead to cirrhosis, end-stage liver disease, liver transplant and even hepatocellular carcinoma. Therefore, the need for treatment for this devastating, progressive disease is dire. That said, the NASH drug development landscape is robust. With over 100 compounds in discovery, nearly 80 drugs in clinical trials in which 4 have made it to Phase III; it is anticipated that a NASH drug will be available within the next few years and there will be several drug categories to boot. Potential drug classes include those that target metabolic pathways such as de novo lipogenesis, those that have anti-inflammatory or anti-apoptotic properties and anti-fibrotic compounds. Similar to the diabetes indication, polypharmacy may be one strategy to tackle this progressive, chronic disease.
Although the NASH pipeline is abundant, with no FDA approved treatment as of yet, there is still time to enter the race. For sponsors considering stepping into the ring, here’s a list of suggestions to navigate the course.
Develop the right plan for your drug.
Demonstrating safety and tolerability in early clinical studies is a must to advance a program forward, but so much more can be captured in these early trials. Pharmacodynamic signals of drug efficacy can be examined in Phase I development through innovative and adaptive study designs which include a patient arm or tailored proof-of-mechanisms studies, like interrogating the de novo lipogenesis pathway for an anti-steatosis drug.
Identify the right participants for your study.
Participant pre-screening efforts can save sponsors time and money. A clinical research organization with a rich database of well characterized participants can expedite study recruitment and reduce screen failures. The FibroScan® is a fast, painless, non-invasive ultrasound-like device that measures liver stiffness (fibrosis) and hepatic steatosis (fat); both measures are key markers for NASH clinical trials. FibroScan® pre-screening can assist with participant selection for more sophisticated and expensive inclusion criteria such as magnetic resonance imaging (MRI) or elastography (MRE). In addition, the Liver Forum, a consortium of academic leaders, industry and regulators, has recently recommended using the FibroScan® as part of the inclusion criteria for early phase NASH clinical studies.
Do more with less.
The FibroScan® is not only a valuable tool for study inclusion criteria, this technique has served as a key primary or secondary study endpoints in many NASH clinical studies. Additionally, soluble biomarker panels such as FIB4 and NAFLD Fibrosis Score calculated from clinical chemistries such as AST and ALT, are also inexpensive ways to identify potential participants for clinical studies and monitor drug efficacy.
Look towards the future.
Current NASH standards of care and management strategies include vitamin E or pioglitazone treatment as these therapies have been shown to be effective in improving the histological features of NASH. Drug-drug interaction studies may be necessary before moving into later phase studies to accommodate patients on standards of care treatment and allowing for a great patient population base to pull from, if safe to do so.
The NASH race to market is well underway; however latecomers entering the NASH indication do not need to fall by the wayside. A strategic early phase drug development plan may help foster the next blockbuster treatment.
Injectable medication can be burdensome for the patient, causing pain and discomfort, and can contribute to compliance issues especially when chronic treatment is required. A promising and emerging area in pharmacology that may alleviate these obstacles is transdermal microneedle drug delivery. Patch microneedles are about the size of a postage stamp and can contain ~30-50 tiny needles smaller than the diameter of a strand of hair. This innovative design limits pain, tissue trauma and infection, and could be applied by minimally-trained personnel, facilitating use in both developed and developing countries.
Patch microneedles are widely gaining traction in the vaccine drug development space. The microneedles penetrate the upper skin layer and rapidly dissolve to deliver the vaccine. Transdermal vaccine delivery is a preferred alternative for those with a fear of needles and is an essential step towards disease eradication worldwide. A critical issue concerning current vaccines reaching rural regions is the lack of refrigeration and a cold supply chain. In this respect, microneedles hold the potential to overcome vaccine wastage due to heat and light exposure as well as volume waste.
This technology is not only being applied to vaccines but across indications from oncology to chronic diseases such as diabetes. Alarmingly, the World Health Organization estimates that only half of all patients with chronic diseases comply with treatment recommendations. Microneedle patches are being developed to address treatment compliance by easing the burden of daily or weekly injections. Through passive drug diffusion, drug delivery could be sustained for 6 months up to 1 year as fixed needles swell within the skin layer to hold the patch in place.
Microneedle patch technology is a promising drug delivery system that may ease patient burden and can facilitate great treatment compliance. A number of current studies in early clinical drug development are underway for a multitude of disease indications. This drug delivery technology has the potential to revolutionize access to treatment, making significant impact on public health worldwide.
Know Your Numbers Campaign
At Celerion, our community outreach does not only serve to inform the public about who we are and medical research opportunities, but we also use this time to give back to our community. In April 2016, Celerion launched a “Know Your Numbers” campaign in the Phoenix, AZ area. Through this initiative, we provide free hemoglobin A1c health checks at local community events.
Hemoglobin A1c reflects blood glucose levels over the past 2 to 3 months. This number helps an individual know if they are in a healthy range; falling in a prediabetes range, indicating risk of developing type 2 diabetes; or if they have diabetes, how well their medication is controlling their glucose levels. In the Phoenix area, the prevalence of type 2 diabetes is estimated at 10.8%, this is considerably higher than the national US average of 9.4%. In addition, the number for prediabetes rates is also startling. According to the CDC, one-in-three people have prediabetes yet only 1 out of 10 know this, making diabetes prevention and awareness essential for our community.
Since the start of this initiative, Celerion has provided 1389 free A1c health checks across 24 events with 14 local community partners. Results from the health checks have been recently published in the journal Trials and shared with our community partners, to aid them in developing strategies to best assist those at risk of developing type 2 diabetes. For our efforts to promote prediabetes and diabetes awareness, Celerion have been recognized by the American Diabetes Association, Maricopa County Health Department, and The Arizona Partnership for Immunization.
References:
National Diabetes Statistics Report, 2017. Estimates of Diabetes and Its Burden in the United States. Atlanta, GA: Centers for Disease Control and Prevention; 2017. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf.
American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33 Suppl 1:S62–9. https://doi.org/10.2337/dc10-S062.
Prediabetes, the New Normal in Early Clinical Research
There is a growing shift in inclusion criteria from healthy normal subjects to patient groups in early drug development. For programs with a chronic metabolic disease investigational product, prediabetes subject enrollment in early phase studies can facilitate the detection of efficacy signals and proof-of-concept [1].
Prediabetes describes a state of glucose impairment which holds a significant risk of developing type 2 diabetes. According to the CDC, 1 in 3 Americans have prediabetes, yet only 1 out of 10 individuals are aware of their condition [2]. Risk factors for prediabetes include being overweight or obese, an estimated 80% of subjects with glucose impairment are overweight or obese [3]. Additional risk factors include 45 years of age or older, a family history of type 2 diabetes, a sedentary lifestyle, and previously having gestational diabetes [2]. Clinical research organizations can provide a helping hand in managing the disease by being committed to raising prediabetes awareness through free A1c screening during community outreach events [4] and offering resources on diabetes prevention.
Type 2 diabetes patients can be vulnerable to hyperglycemic and hypoglycemic events during early phase placebo-controlled studies, monotherapy studies or during washout periods. Since prediabetes subjects maintain a degree of glucose control, they are less likely to experience severe glucose excursions. Therefore, a risk mitigation strategy to minimize serious glycemic changes in early dose escalation studies could include the enrollment of prediabetes subjects. In addition, to better understand a drug’s effect on glycemic parameters, intensive daily glucose tracking through continuous glucose monitoring (CGM) can provide blood glucose readings every 5-15 minutes for up to 2 weeks. To this end, CGM is recognized as the “ECG Holter-monitor for glycemia” [5].
Nonalcoholic fatty liver disease (NAFLD) is the next major health epidemic and prediabetes is strongly associated with hepatic inflammation and fibrosis in NAFLD subjects [6]. Nonalcoholic steatohepatitis (NASH), a deleterious form of the disease can lead to liver cirrhosis, end-stage liver failure and even hepatocellular carcinoma. Using non-invasive techniques to assess steatosis, inflammation and oxidative stress, and fibrosis can expedite clinical research. These pharmacodynamic imaging and soluble biomarker analyses are essential early signals of drug efficacy to enable faster go/no-go decisions.
Prediabetes subject enrollment in early clinical studies for weight management, Type 2 Diabetes or NASH indications offers an opportunity to examine pharmacodynamic endpoints in a population of interest while minimizing safety concerns typically observed with patient studies.
5. Vigersky R, Shrivastav M. Role of continuous glucose monitoring for type 2 in diabetes management and research. J Diabetes Complications. 2017;31(1):280-7.
6. Yilmaz Y, Senates E, Yesil A, Ergelen R, Colak Y. Not only type 2 diabetes but also prediabetes is associated with portal inflammation and fibrosis in patients with non-alcoholic fatty liver disease. J Diabetes Complications. 2014;28(3):328-31.
Supporting the Next Major Health Epidemic
Non-alcoholic fatty liver disease (NAFLD)/Non-alcoholic steatohepatitis (NASH) is viewed as the next major health epidemic and with no current anti-NASH medication on the market to treat this chronic disease; it is expected to place a heavy burden on our healthcare systems.
NAFLD is considered to be a hepatic manifestation of metabolic syndrome, associated with obesity and insulin resistance. The incidence of NAFLD/NASH is rapidly increasing worldwide, currently affecting over 1 billion people, and the prevalence of NAFLD/NASH is not limited to adults as the rise of this disease is also observed in children. Presently, the standard of care for NAFLD/NASH includes invasive liver biopsy procedures for diagnosis and management. Therefore, NASH is expected to become the most common indication for liver transplantation in the United States by 2020, surpassing viral diseases like hepatitis C.
This month, Thomas Jefferson University Hospital in Philadelphia reported its 1,000th liver transplant surgery. This milestone calls out to our industry the importance of our work in support of patients with end-stage liver disease.
Non-invasive diagnostic and prognostic techniques to identify NASH patients and those who will positively response to a given treatment regimen is overwhelmingly needed. Recent advancements in medical imaging, such as magnetic resonance elastography and FibroScan, are contributing to achieving these goals. In addition, an emerging trend in early clinical research is to examine pharmacodynamic effects in patient populations. In this respect, imaging tools as well as soluble biomarkers found in bodily fluids such as blood, urine, saliva, etc. are extremely useful to evaluate NAFLD/NASH drug efficacy.
A number of pharmaceutical companies are currently involved in NAFLD/NASH research. This list includes, but is not limited to Intercept, Genfit, Galmed, Gilead, Genentech, Pfizer, Merck, and Novartis.
An experienced contract research organization (CRO) will have NAFLD/NASH capabilities and experience in hepatic lipid metabolism and fibrosis biomarkers to incorporate into drug development Single Ascending Dose (SAD), Multi-Ascending Dose (MAD), Drug Interaction (DDI), Phase 1b, Proof-of-Concept (POC) studies. The scientific and clinical operational staff will have experience and thorough understanding of the disease as well as bioanalytical and non-invasive measurements of hepatic lipid metabolism, inflammation and fibrosis. In addition, the CRO will have a strong recruitment track-record to support special-population studies like diabetes, obesity, NAFLD programs.
Celerion’s experienced and knowledgeable scientific and clinical operational team can help navigate NAFLD/NASH drug development programs through this emerging market.
Science leading to medicine, done well, helps saves patients’ lives and provides for a healthy standard of living.
Celerion was recognized by Biopharmaceutical Clients in May 2016 as a Leader in Contract Research Quality Benchmarking Survey.
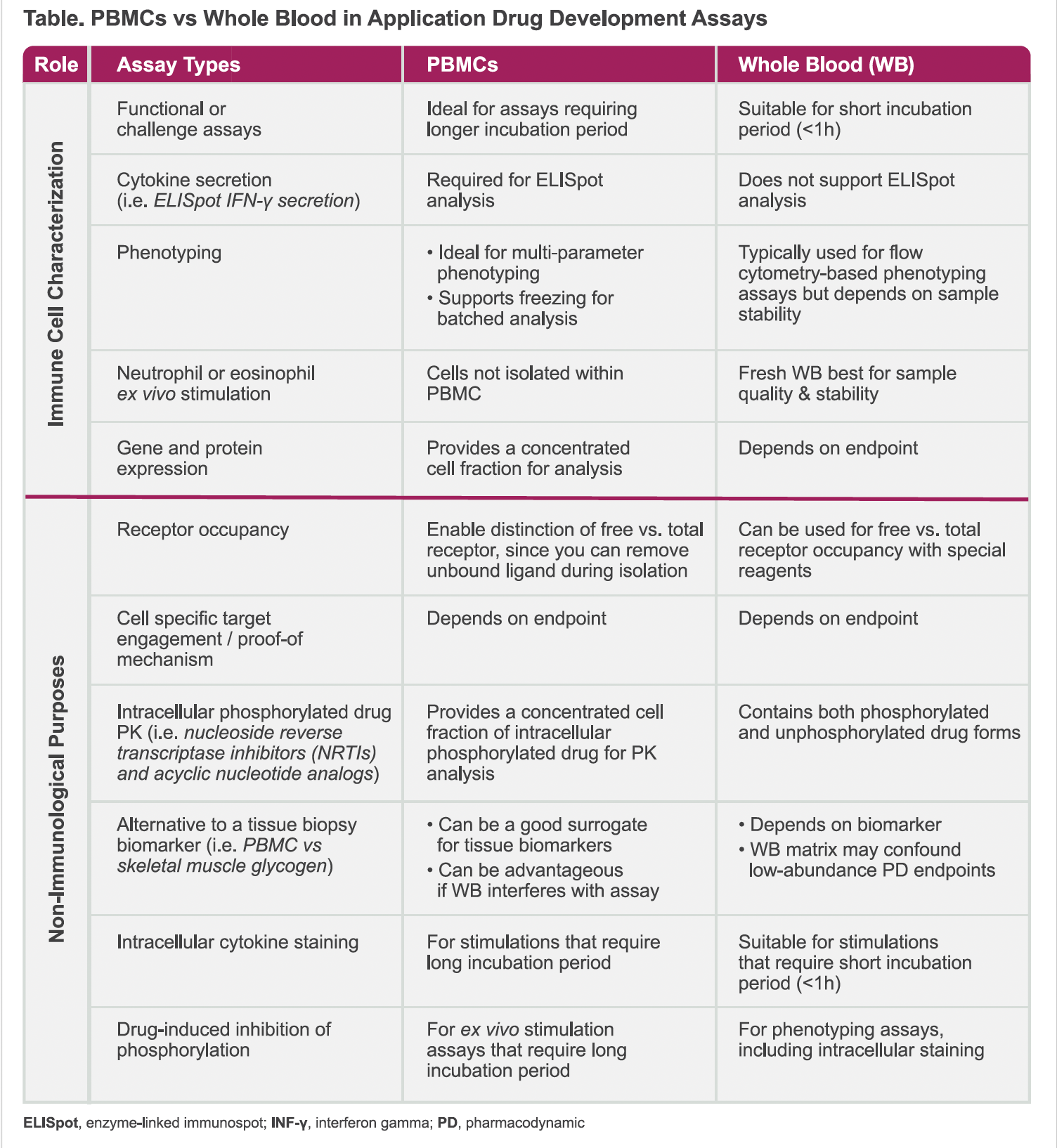
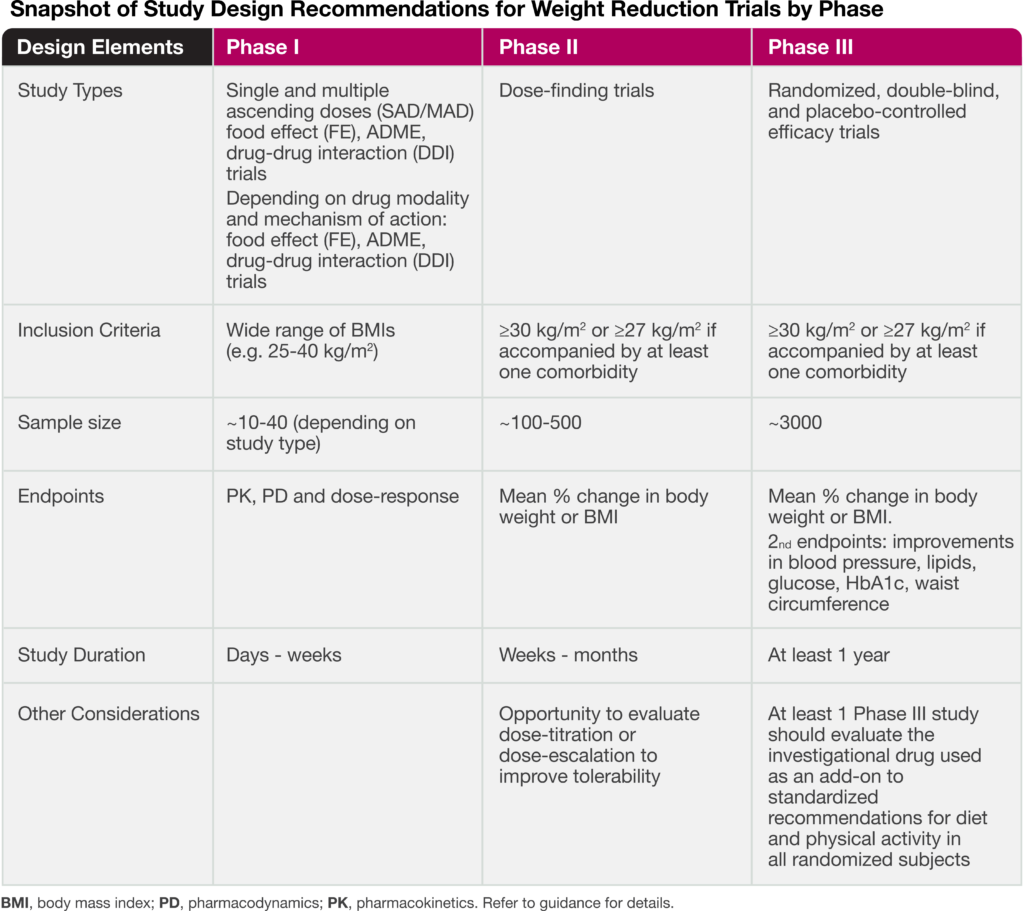

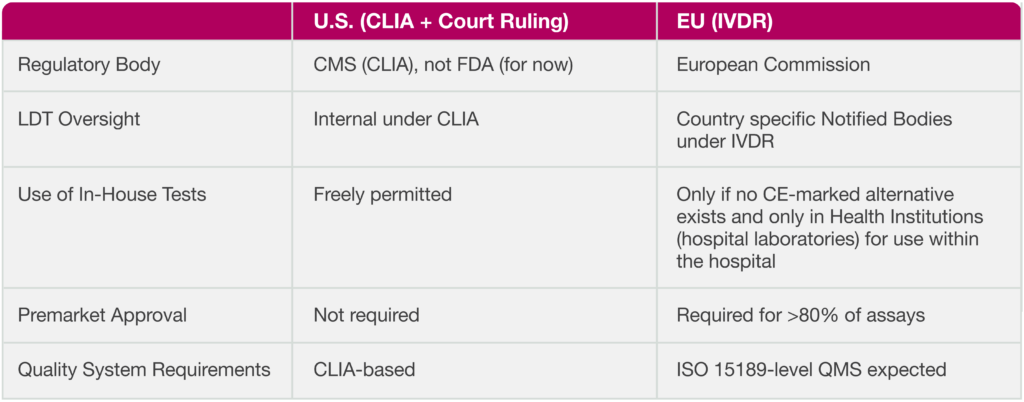

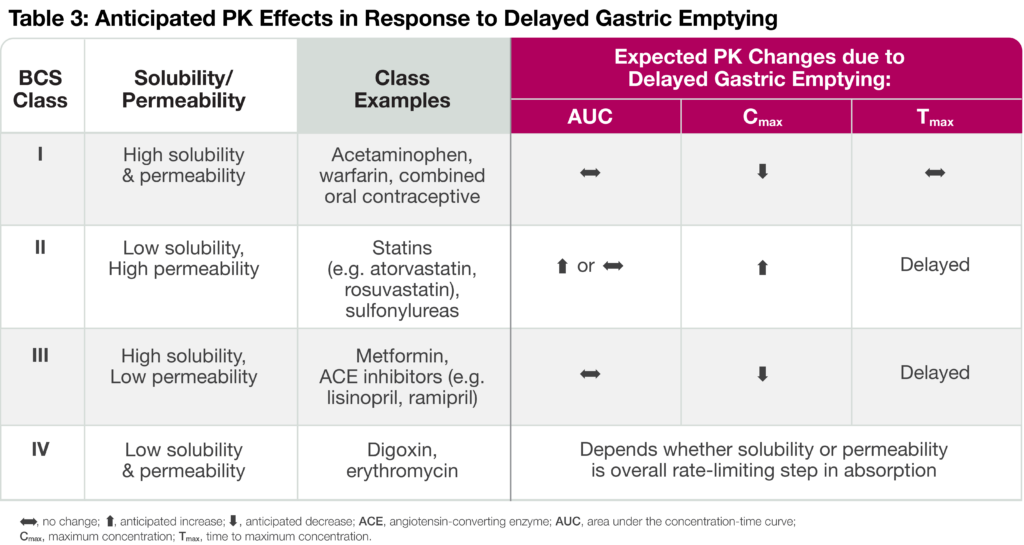

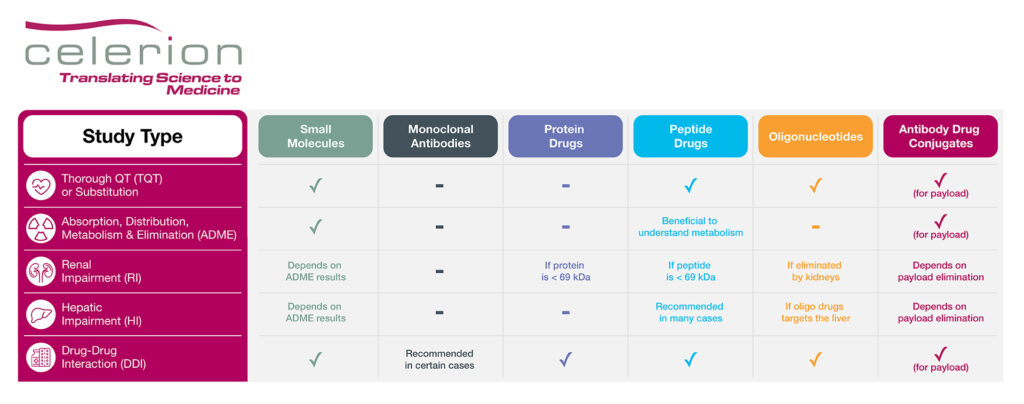
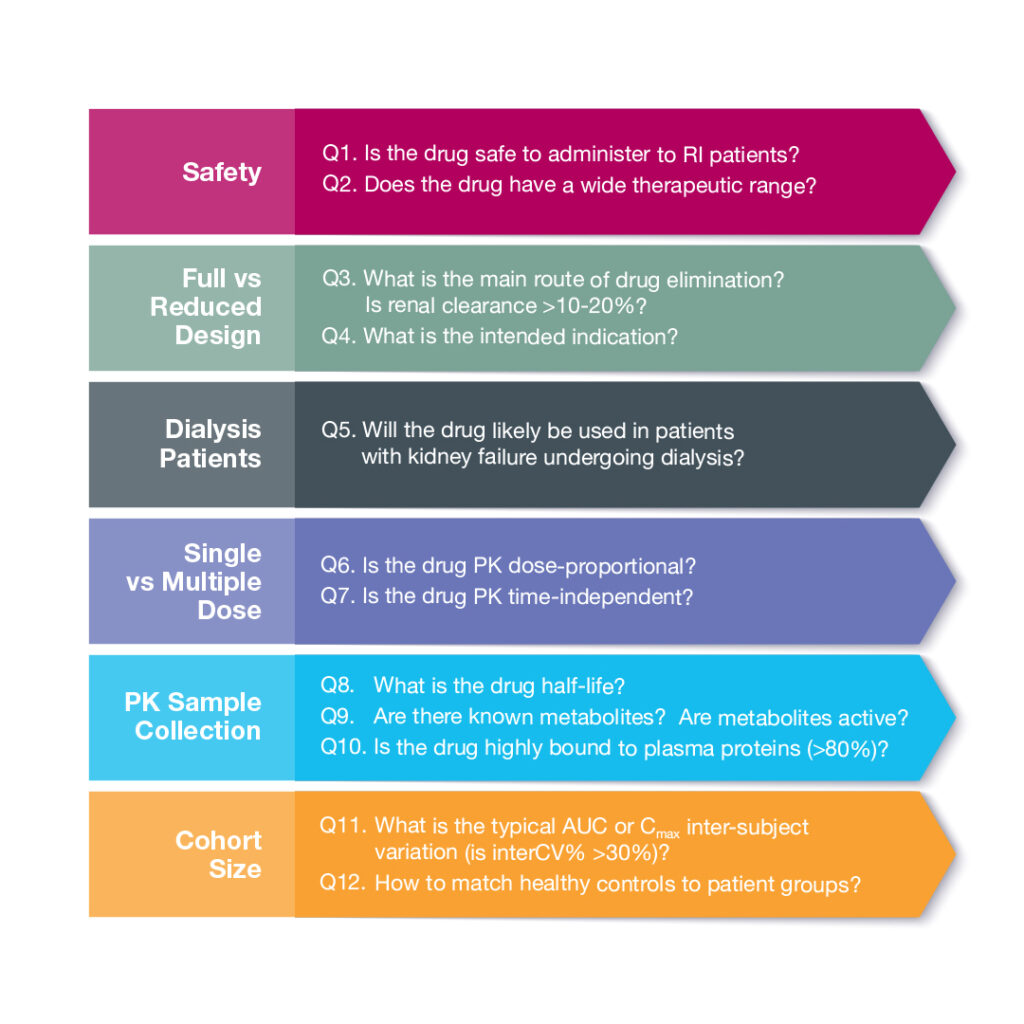


 by Sabina Paglialunga, PhD – Director, Scientific Affairs, Celerion
by Sabina Paglialunga, PhD – Director, Scientific Affairs, Celerion


 by Sabina Paglialunga, PhD – Director, Scientific Affairs, Celerion
by Sabina Paglialunga, PhD – Director, Scientific Affairs, Celerion By Sumit Kar, Lead Scientist – Biomarkers, Celerion
By Sumit Kar, Lead Scientist – Biomarkers, Celerion